Overview: The WGCNA package (in R) uses functions that perform a correlation network analysis of large, high-dimensional data sets (RNAseq datasets). This unbiased approach clusters similarly expressed genes into groups (termed 'modules') which are then correlated with quantitative or categorical traits measured in the experiment. Modules can be further analyzed using GO, KEGG or KOG, VisANT and Cytoscape. This approach goes beyond a simple 'list of genes' and helps tease apart large, complicated RNAseq datasets into functional clusters that are more easy to interpret.
Figure 1: Overview of WGCNA package

Lecture: WGCNA Concepts
R script: WGCNAshortTutorial.R
Get set up for the exercise:
When following along here, please switch to your idev session for running these example commands.
If you have not requested an idev session, do so now:
ssh <username>@stampede2.tacc.utexas.edu idev -m 120 -q normal -A UT-2015-05-18 -r RNASeq-Thu
#We will be doing all this in the idev session cds cd my_rnaseq_course/day_4_partA/wgcna
Explanation of sample dataset: Time series of coral larval development from 4 hours post fertilization (Day 0) to 245 hours post fertilization (Day 12). Multiple other quantitative traits were measured through the time series. Only green and red fluorescence are added as quantitative traits in the sample dataset. Dataset has 48 samples total, four replicates (A-D) over 12 days. The goal is to find genes that correlate with developmental traits through time and differences in gene expression between early larval development and late larval development.
The complete R script has been provided for you, so you run it using R CMD BATCH. Or you can open up an R prompt and run key pieces of it by copy-pasting bits of code from below. This is to understand what the code is actually doing.
TRAIT DATA FILE: Traits_23May2015.csv
module load intel/18.0.2 module load Rstats/4.0.3
module load biocontainers module load r-flashclust/ctr-1.01_2--r3.3.2_0 module load r-wgcna/ctr-1.51--r3.3.2_0
R CMD BATCH WGCNAshortTutorial.R
Below are the details of the R code you just kicked off above. NO NEED TO RUN THESE LINE BY LINE!
Step 1: upload data into R and reformat for WGCNA (This is all run under the R console)
# Only run the following commands once to install flashClust if needed
#install.packages("flashClust")
# Load WGCNA and flashClust libraries every time you open R
library(WGCNA)
library(flashClust)
# Uploading data into R and formatting it for WGCNA
# This creates an object called "datExpr" that contains the normalized counts file output from DESeq2
datExpr = read.csv("SampleTimeSeriesRLD.csv")
# "head" the file to preview it
head(datExpr) # You see that genes are listed in a column named "X" and samples are in columns
# Manipulate file so it matches the format WGCNA needs
row.names(datExpr) = datExpr$X
datExpr$X = NULL
datExpr = as.data.frame(t(datExpr)) # now samples are rows and genes are columns
dim(datExpr) # 48 samples and 1000 genes (you will have many more genes in reality)
# Run this to check if there are gene outliers
gsg = goodSamplesGenes(datExpr, verbose = 3)
gsg$allOK
#If the last statement returns TRUE, all genes have passed the cuts. If not, we remove the offending genes and samples from the data with the following:
#if (!gsg$allOK)
# {if (sum(!gsg$goodGenes)>0)
# printFlush(paste("Removing genes:", paste(names(datExpr)[!gsg$goodGenes], collapse= ", ")));
# if (sum(!gsg$goodSamples)>0)
# printFlush(paste("Removing samples:", paste(rownames(datExpr)[!gsg$goodSamples], collapse=", ")))
# datExpr= datExpr[gsg$goodSamples, gsg$goodGenes]
# }
#Create an object called "datTraits" that contains your trait data
datTraits = read.csv("Traits_23May2015.csv")
head(datTraits)
#form a data frame analogous to expression data that will hold the clinical traits.
rownames(datTraits) = datTraits$Sample
datTraits$Sample = NULL
table(rownames(datTraits)==rownames(datExpr)) #should return TRUE if datasets align correctly, otherwise your names are out of order
head(datTraits)
# You have finished uploading and formatting expression and trait data
# Expression data is in datExpr, corresponding traits are datTraits
save(datExpr, datTraits, file="SamplesAndTraits.RData")
#load("SamplesAndTraits.RData")
At this point you will need to identify sample outliers and choose a soft threshold power. These are easy to do and are well documented in the online tutorials. It is suggested that you cluster samples by expression to identify any outliers before this step. This is provided in the attached R script.
# Choose a soft threshold power- USE A SUPERCOMPUTER IRL ------------------------------------
powers = c(c(1:10), seq(from =10, to=30, by=1)) #choosing a set of soft-thresholding powers
sft = pickSoftThreshold(datExpr, powerVector=powers, verbose =5, networkType="signed") #call network topology analysis function
sizeGrWindow(9,5)
par(mfrow= c(1,2))
cex1=0.9
plot(sft$fitIndices[,1], -sign(sft$fitIndices[,3])*sft$fitIndices[,2], xlab= "Soft Threshold (power)", ylab="Scale Free Topology Model Fit, signed R^2", type= "n", main= paste("Scale independence"))
text(sft$fitIndices[,1], -sign(sft$fitIndices[,3])*sft$fitIndices[,2], labels=powers, cex=cex1, col="red")
abline(h=0.90, col="red")
plot(sft$fitIndices[,1], sft$fitIndices[,5], xlab= "Soft Threshold (power)", ylab="Mean Connectivity", type="n", main = paste("Mean connectivity"))
text(sft$fitIndices[,1], sft$fitIndices[,5], labels=powers, cex=cex1, col="red")
#from this plot, we would choose a power of 18 becuase it's the lowest power for which the scale free topology index reaches 0.90
Figure 2: Soft Thresholding: from this plot, we would choose a power of 18 since it's the lowest power for which the scale free topology index reaches 0.90 (red line)

Step 2: Construct a gene co-expression network and identify modules
#build a adjacency "correlation" matrix
enableWGCNAThreads()
softPower = 18
adjacency = adjacency(datExpr, power = softPower, type = "signed") #specify network type
head(adjacency)
# Construct Networks- USE A SUPERCOMPUTER IRL -----------------------------
#translate the adjacency into topological overlap matrix and calculate the corresponding dissimilarity:
TOM = TOMsimilarity(adjacency, TOMType="signed") # specify network type
dissTOM = 1-TOM
# Generate Modules --------------------------------------------------------
# Generate a clustered gene tree
geneTree = flashClust(as.dist(dissTOM), method="average")
plot(geneTree, xlab="", sub="", main= "Gene Clustering on TOM-based dissimilarity", labels= FALSE, hang=0.04)
#This sets the minimum number of genes to cluster into a module
minModuleSize = 30
dynamicMods = cutreeDynamic(dendro= geneTree, distM= dissTOM, deepSplit=2, pamRespectsDendro= FALSE, minClusterSize = minModuleSize)
dynamicColors= labels2colors(dynamicMods)
MEList= moduleEigengenes(datExpr, colors= dynamicColors,softPower = softPower)
MEs= MEList$eigengenes
MEDiss= 1-cor(MEs)
METree= flashClust(as.dist(MEDiss), method= "average")
save(dynamicMods, MEList, MEs, MEDiss, METree, file= "Network_allSamples_signed_RLDfiltered.RData")
#plots tree showing how the eigengenes cluster together
#INCLUE THE NEXT LINE TO SAVE TO FILE
#pdf(file="clusterwithoutmodulecolors.pdf")
plot(METree, main= "Clustering of module eigengenes", xlab= "", sub= "")
#set a threhold for merging modules. In this example we are not merging so MEDissThres=0.0
MEDissThres = 0.0
merge = mergeCloseModules(datExpr, dynamicColors, cutHeight= MEDissThres, verbose =3)
mergedColors = merge$colors
mergedMEs = merge$newMEs
#INCLUE THE NEXT LINE TO SAVE TO FILE
#dev.off()
#plot dendrogram with module colors below it
#INCLUE THE NEXT LINE TO SAVE TO FILE
#pdf(file="cluster.pdf")
plotDendroAndColors(geneTree, cbind(dynamicColors, mergedColors), c("Dynamic Tree Cut", "Merged dynamic"), dendroLabels= FALSE, hang=0.03, addGuide= TRUE, guideHang=0.05)
moduleColors = mergedColors
colorOrder = c("grey", standardColors(50))
moduleLabels = match(moduleColors, colorOrder)-1
MEs = mergedMEs
#INCLUE THE NEXT LINE TO SAVE TO FILE
#dev.off()
save(MEs, moduleLabels, moduleColors, geneTree, file= "Network_allSamples_signed_nomerge_RLDfiltered.RData")
Figure 3: Clustering dendrogram of all genes, with dissimilarities based on topological overlap. Each vertical line represents a single gene. Assigned module colors below.
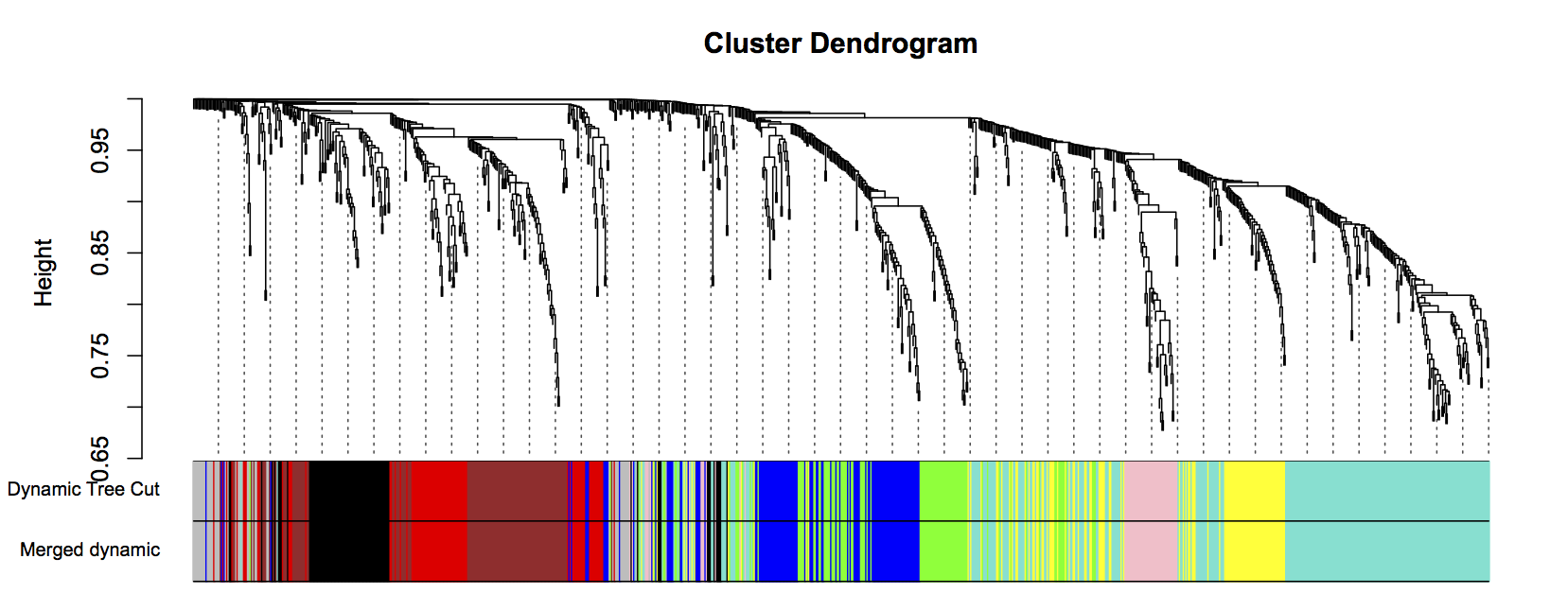
Step 3: Relate modules to external traits
# Correlate traits --------------------------------------------------------
#Define number of genes and samples
nGenes = ncol(datExpr)
nSamples = nrow(datExpr)
#Recalculate MEs with color labels
MEs0 = moduleEigengenes(datExpr, moduleColors)$eigengenes
MEs = orderMEs(MEs0)
moduleTraitCor = cor(MEs, datTraits, use= "p")
moduleTraitPvalue = corPvalueStudent(moduleTraitCor, nSamples)
#Print correlation heatmap between modules and traits
textMatrix= paste(signif(moduleTraitCor, 2), "\n(",
signif(moduleTraitPvalue, 1), ")", sep= "")
dim(textMatrix)= dim(moduleTraitCor)
par(mar= c(6, 8.5, 3, 3))
#display the corelation values with a heatmap plot
#INCLUE THE NEXT LINE TO SAVE TO FILE
#pdf(file="heatmap.pdf")
labeledHeatmap(Matrix= moduleTraitCor,
xLabels= names(datTraits),
yLabels= names(MEs),
ySymbols= names(MEs),
colorLabels= FALSE,
colors= blueWhiteRed(50),
textMatrix= textMatrix,
setStdMargins= FALSE,
cex.text= 0.5,
zlim= c(-1,1),
main= paste("Module-trait relationships"))
#INCLUE THE NEXT LINE TO SAVE TO FILE
#dev.off()
Figure 4: Module-Trait relationships. Color scale (red-blue) represents the strength of the correlation between the module and the trait. For example, the turquoise module is highly significantly correlated with HPF, RedFluoro and GreenFluoro. Each box gives a correlation value (R^2) followed by p-value (in parenthesis).

For further analysis, if you wanted to pull out genes belonging to a certain module, you can use the following command: