The first order of business after receiving sequencing data should be to check your data quality. This often-overlooked step helps guide the manner in which you process the data, and can prevent many headaches.
FastQC
FastQC is a tool that produces a quality analysis report on FASTQ files.
Useful links:
- FastQC report for a good Illumina dataset
- FastQC report for a bad Illumina dataset
- Online documentation for each FastQC report
First and foremost, the FastQC "Summary" should generally be ignored. Its "grading scale" (green - good, yellow - warning, red - failed) incorporates assumptions for a particular kind of experiment, and is not applicable to most real-world data. Instead, look through the individual reports and evaluate them according to your experiment type.
The FastQC reports I find most useful are:
- The Per base sequence quality report, which can help you decide if sequence trimming is needed before alignment.

2. The Per Sequence Quality Score report, which can tell you if a subset of your reads just have poor quality scores. These reads can be completely filtered from analysis.

3. The Sequence Duplication Levels report, which helps you evaluate library enrichment / complexity. But note that different experiment types are expected to have vastly different duplication profiles.

- The Overrepresented Sequences report, which helps evaluate adapter contamination.
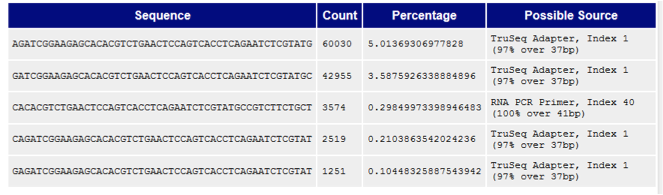
Note: For many of its reports, FastQC analyzes only the first 200,000 sequences in order to keep processing and memory requirements down.
Running FastQC
FastQC iis available on lonestar as a module.
Here's how to run FastQC on our sample data:
module load fastqc fastqc data/Sample1_R1.fastq
Exercise: FastQC results
What did FastQC create?
Looking at FastQC output
You can't run a web browser directly from your "dumb terminal" command line environment. The FastQC results have to be placed where a web browser can access them. We put a copy at this URL:
http://web.corral.tacc.utexas.edu/BioITeam/rnaseq_course/fastqc_exercise/Sample1_R1_fastqc/fastqc_report.html
Exercise: Should we trim this data?
Based on this FastQC output, should we trim this data?
Let's look at tools to do such manipulations to fastqc files, if we have to.