{kind=link}
Overview
As has been mentioned several times, variants are anything that is different from a reference genome, but large insertions of novel DNA represent something of an unknown unknown. If a read doesn't map is it because its some kind of contamination, or is it because something new has entered into the sample. This tutorial is an attempt to use tools you are already familiar with to identify such novel DNA mutations. As this is a new tutorial any feedback is very welcome and helpful.
Prerequisite required
This tutorial assumes you have already installed spades in the assembly tutorial. Verify you have done so with the 'spades.py -v' command does not return 3.13.0, need to at least complete enough of that tutorial that spades is installed.
Learning Objectives
- Extract reads where one or both reads do not map to reference
- de novo assemble reads
Relationship to other tutorials
- As presence of adapter can cause problems with assembly, make sure adapters have been trimmed
- Reads will be mapped with bowtie2
- non-mapped reads will be extracted, consider checking quality of these reads and possible additional trimming
- Reads will then be assembled
None of these other tutorials are required to complete this tutorial, but additional information about individual steps may be found there.
Identification of a novel plasmid
One example of novel DNA being present is when a given sample may have a virus or plasmid associated with a sample. Here we will take a sample known to have a high copy plasmid associated with it, but map the reads against only the genome. Unaligned reads would then be expected to be able to assemble into a plasmid.
Get some data
mkdir $SCRATCH/GVA_novel_DNA cd $SCRATCH/GVA_novel_DNA cp $BI/gva_course/novel_DNA/* . ls
^ above transfers 2 fastq files and a reference file.
Map read using bowtie2
This is the same process used in the read mapping tutorial, and therefore presented with little comment except to remark on differences. Refer to that tutorial for more in-depth information.
- Convert reference to fasta
module load bioperl bp_seqconvert.pl --from genbank --to fasta < CP009273.1_Eco_BW25113.gbk > CP009273.1_Eco_BW25113.fasta
Remember to make sure you are on an idev done
For reasons discussed numerous times throughout the course already, please be sure you are on an idev done. It is unlikely that you are currently on an idev node as copying the files while on an idev node seems to be having problems as discussed. Remember the hostname command and showq -u can be used to check if you are on one of the login nodes or one of the compute nodes. If you need more information or help re-launching a new idev node, please see this tutorial.
Index the referencemkdir bowtie2 module load bowtie/2.3.4 bowtie2-build CP009273.1_Eco_BW25113.fasta bowtie2/CP009273.1_Eco_BW25113
The following command will take ~5 minutes to complete. Before you run the command execute '
bowtie2 -h' so while the command is running you can try to figure out what the different options are doing that we did not include in our first tutorial.Map readsbowtie2 --very-sensitive-local -t -p 48 -x bowtie2/CP009273.1_Eco_BW25113 -1 SRR4341249_1.fastq -2 SRR4341249_2.fastq -S bowtie2/SRR4341249-vsl.sam --un-conc SRR4341249-unmapped-vsl.fastq
Assemble unmapped reads
This is the same process used in the plasmid assembly portion of the genome assembly tutorial, and therefore presented with little comment except to remark on differences. Refer to that tutorial for more in-depth information.
Attempt to assemble with plasmidspades.py which expects to find circularized plasmid type sequences. Command expected to take ~5 minutes
run plasmid spadesplasmidspades.py -t 48 -o unmapped_plasmid -1 SRR4341249-unmapped-vsl.1.fastq -2 SRR4341249-unmapped-vsl.2.fastq
Additionally attempt to assemble with base spades.py command which uses different internal settings to potentially identify different types of novel DNA from our unmapped reads. Command expected to take ~5 minutes.
run plasmid spadesspades.py -t 48 -o unmapped_full -1 SRR4341249-unmapped-vsl.1.fastq -2 SRR4341249-unmapped-vsl.2.fastq
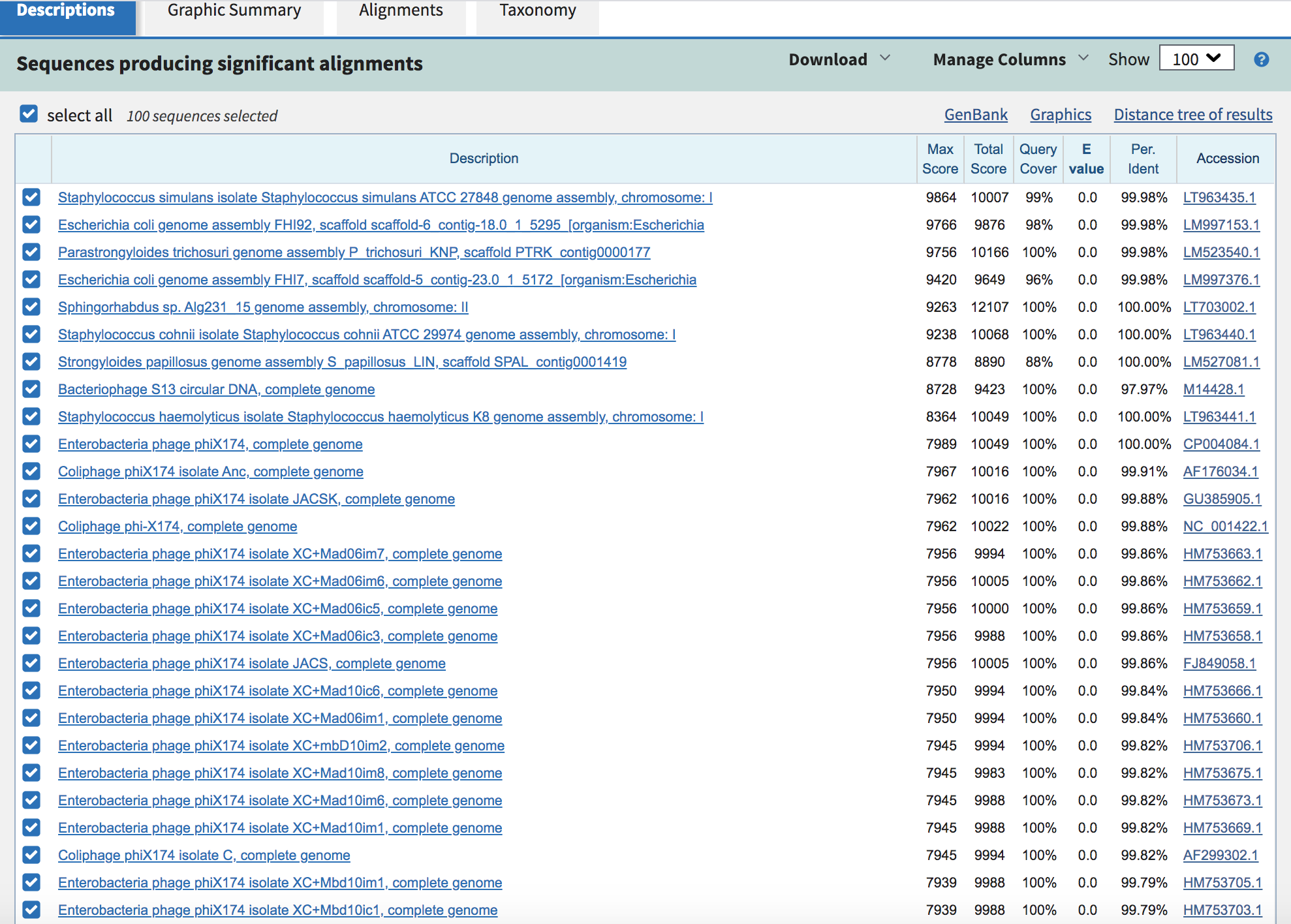
Compare contigs generated from different assemblies
Recall from genome assembly tutorial the file we are most interested in is the contigs.fasta file in each output directory (unmapped_plasmid and unmapped_full).
Next steps
Here we have presented a proof of concept that unmapped reads can be used to find something that we actually did know was present. We also found something that was even longer that wasn't expected.
Additional questions are:
{kind=link}