Demultiplexing 2bRAD data exercise:
This exercise should be done on TACC.
First copy over the exercise directory and prepare reads:
#start an idev session if you have not already idev #go to coure directory on scratch cds cd rad_intro/ #copy over the demultiplexing directory cp -r /work/02260/grovesd/lonestar/intro_to_rad_2017/demultiplexing/process_2bRAD . cd process_2bRAD ls -l *.gz #returns these files: A_CTGCAG_L004_R1_001.fastq.gz A_CTGCAG_L005_R1_001.fastq.gz B_GAAGTT_L004_R1_001.fastq.gz B_GAAGTT_L005_R1_001.fastq.gz #A few things to notice about these files: #They are compressed with gzip (indicated by .gz) #We have two groups, A and B, that are labeled with #barcode sequences CTGCAG and GAAGTT respectively. #These groups represent pools of samples that all had #the same barcode (attached during PCR step in library prep) #For each group A and B, there are two files, L004 and L005. #These files came from two lanes on the Illumina sequencing run, #But represent the same groups of samples, so we will concatenate them. #Finally, note that all four fastq files have 'R1' #Unlike in the ddRAD example, there are no R2s #Because 2bRAD reads are short (32-36bp), there is not reason #to do paired end sequencing. #In summary we have single-end read data from two pools of samples sequenced across two lanes #decompress the files gunzip *.gz #concatenate the lane 4 and lane 5 data for each pool cat A_CTGCAG_L004_R1_001.fastq A_CTGCAG_L005_R1_001.fastq > A_CTGCAG_R1.fastq cat B_GAAGTT_L004_R1_001.fastq B_GAAGTT_L005_R1_001.fastq > B_GAAGTT_R1.fastq #Look at the barcode data to get an idea of what these fastq files are #This is the same information that would be uploaded to GSAF when the #samples were submitted for sequencing. less barcode_data.tsv
The same barcodes information is here
Compare this with expected final product from library prep:
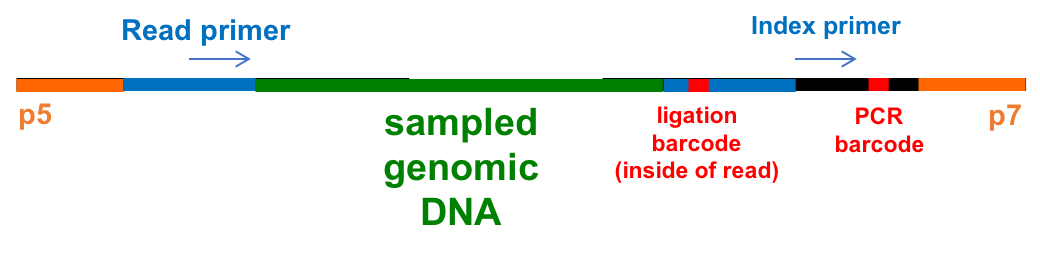
Within each group (A or B) all samples had the same PCR barcode which appears in the file name (6 bases long).
The PCR barcode was read by the indexing primer and will not show up in the reads themselves. GSAF already used this barcode to demultiplex the reads for you into the groups A and B.
The individual samples in these groups have different ligation barcodes (4 bases long), which will appear within the sequencing reads. These ligation barcodes will be used to demultiplex the pooled fastq files for groups A and B into fastqs for the individual samples.
bcgl cut site:

#This library was prepared with bcgl restriction enzyme #Check that the cut site appears in the reads #How many reads do we have? expr $(cat A_CTGCAG_R1.fastq | wc -l) / 4 expr $(cat B_GAAGTT_R1.fastq | wc -l) / 4 #7011846 #(Note these are for demo purposes and shorter than they should be, and would not have the same number of reads in real life) #How many of the reads have the cut site? grep "CGA......TGC" A_CTGCAG_R1.fastq | wc -l #3429158 #3429158/7011846 = 0.49 #Why is the cut site only in half the reads!?
Solution:
Demultiplex the reads:
#The command to run trim2bRAD_2barcodes_dedup.pl is complicated
#Another script -- 2bRAD_trim_launch_dedup.pl -- will generate the command for us:
2bRAD_trim_launch_dedup.pl R1.fastq
#returns our next commands to execute:
trim2bRAD_2barcodes_dedup.pl input=A_CTGCAG_R1.fastq site=".{12}CGA.{6}TGC.{12}|.{12}GCA.{6}TCG.{12}" adaptor="AGATC" sampleID=100
trim2bRAD_2barcodes_dedup.pl input=B_GAAGTT_R1.fastq site=".{12}CGA.{6}TGC.{12}|.{12}GCA.{6}TCG.{12}" adaptor="AGATC" sampleID=100
Check results:
#The resulting files are labled with their barcode combinations and a *tr0 suffix ls -l A*tr0 ls -l B*tr0 #How many did we get? ls -l A*tr0 | wc -l ls -l B*tr0 | wc -l #Looking back at our barcodes, does this make sense? #Which sample does B_GAAGTT_R1_ACCA.tr0 come from? #search for a line in the barcode file with both the barcodes grep TGGT barcode_data.tsv | grep GAAGTT #MCNA13 #Why search for TGGT instead of ACCA? #The ligation barcode on 3Illbc was TGGT, but was then read as ACCA, so that's what shows up in the read