| Table of Contents | ||
|---|---|---|
|
Overview
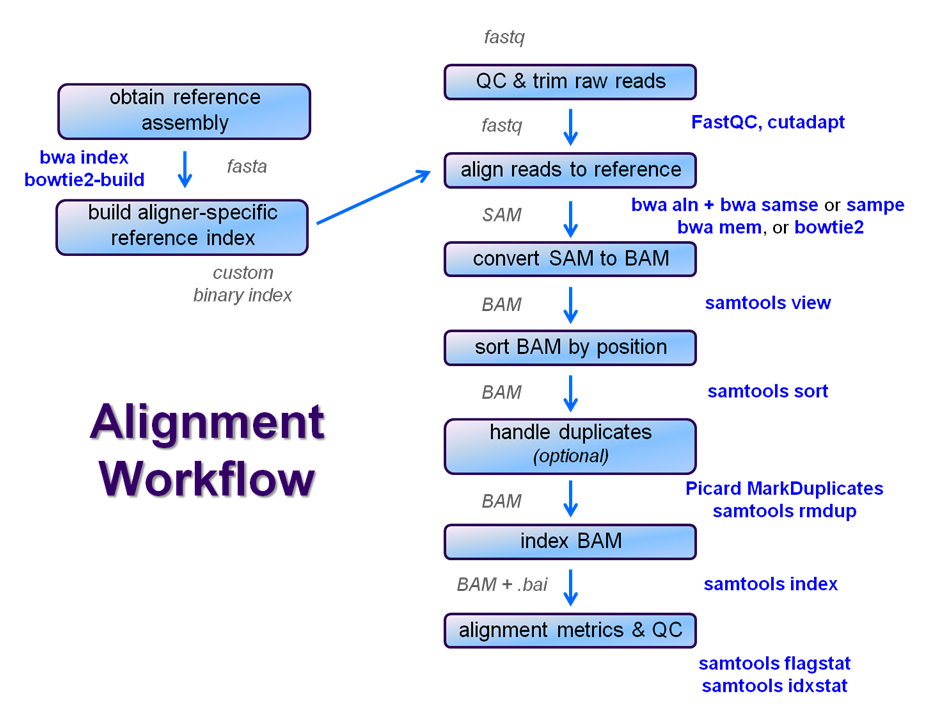
After raw sequence files are generated (in FASTQ format), quality-checked, and pre-processed in some way, the next step in most NGS pipelines is mapping to a reference genome.
For individual sequences, it is common to use a tool like BLAST to identify genes or species of origin. However, a normal NGS dataset will have tens to hundreds of millions of sequences, which BLAST and similar tools are not designed to handle. Thus, a large set of computational tools have been developed to quickly, and with sufficient (but not absolute - and this tradeoff is an important consideration when constructing alignment pipelines) accuracy align each read to its best location, if any, in a reference.
Even though many mapping tools exist, a few individual programs have a dominant "market share" of the NGS world. These programs vary widely in their design, inputs, outputs, and applications. In this section, we will primarily focus on two of the most versatile mappers: BWA and Bowtie2, the latter being part of the Tuxedo suite (e.g. which includes the transcriptome-aware Tophat2) which also includes tools for manipulating NGS data after alignmentRNA-seq aligner Tophat2 as well as other downstream quantifiaction tools.
Connect to login5.ls5.tacc.utexas.edu
...