...
| Tip |
|---|
|
Use our summer school reservation (CoreNGSday4CoreNGS-Thu) when submitting batch jobs to get higher priority on the ls6 normal queue today: sbatch --reservation=CoreNGSday4 CoreNGS-Thu <batch_file>.slurm
idev -m 180 -N 1 -A OTH21164 -r CoreNGSday4CoreNGS-Thu
|
Overview
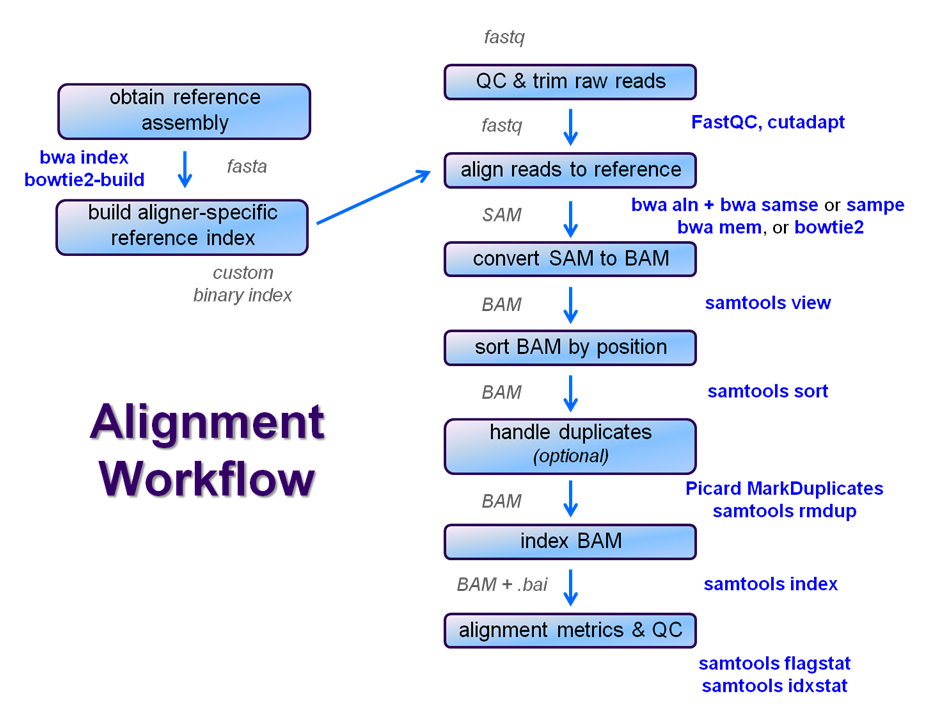 Image Removed
Image Removed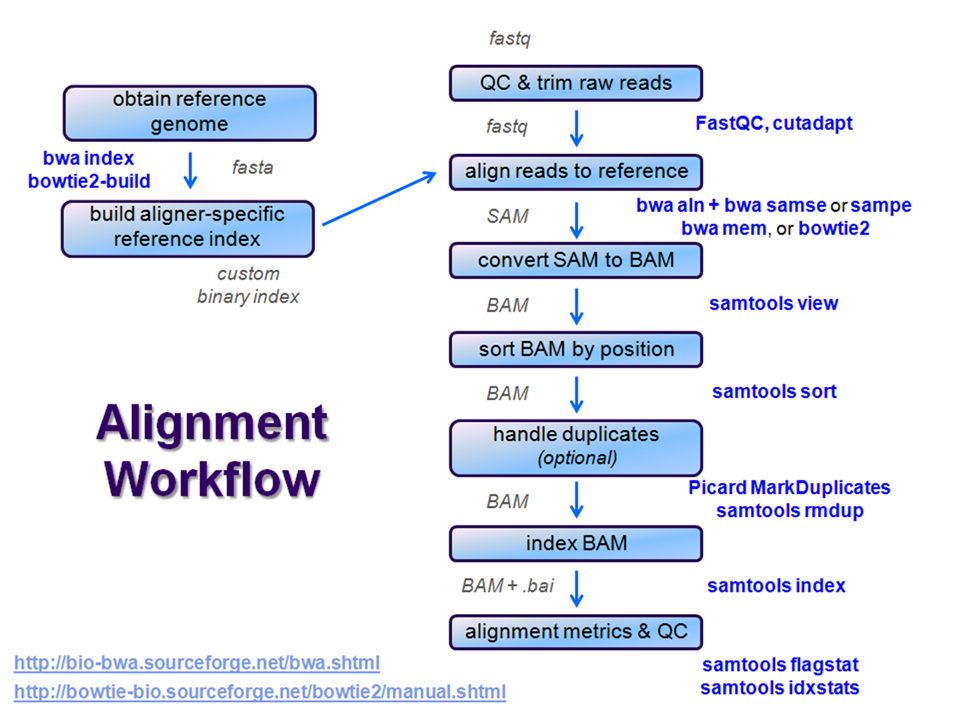 Image Added
Image Added
After raw sequence files are generated (in FASTQ format), quality-checked, and pre-processed in some way, the next step in many NGS pipelines is mapping to a reference genome.
...
Even though many mapping tools exist, a few individual programs have a dominant "market share" of the NGS world. In this section, we will primarily focus on two of the most versatile general-purpose ones: BWA and Bowtie2 (the latter being part of the Tuxedo suite which includes the transcriptome-aware RNA-seq aligner Tophat2 as well as other downstream quantifiaction quantification tools).
Stage the alignment data
...
| Code Block |
|---|
| language | bash |
|---|
| title | Start an idev session |
|---|
|
idev -m 180 -N 1 -A OTH21164 -r CoreNGSday4CoreNGS-Thu |
Then stage the sample datasets and references we will use.
| Code Block |
|---|
| language | bash |
|---|
| title | Get the alignment exercises files |
|---|
|
# Copy the FASTA files for building references
mkdir -p $SCRATCH/core_ngs/references/fasta
cp $CORENGS/references/fasta/*.fa $SCRATCH/core_ngs/references/fasta/
# Copy the FASTQ files that will be used for alignment
mkdir -p $SCRATCH/core_ngs/alignment/fastq
cp $CORENGS/alignment/*fastq.gz $SCRATCH/core_ngs/alignment/fastq/
cd $SCRATCH/core_ngs/alignment/fastq |
...
| File Name | Description | Sample |
|---|
| Sample_Yeast_L005_R1.cat.fastq.gz | Paired-end Illumina, First of pair, FASTQ | Yeast ChIP-seq |
| Sample_Yeast_L005_R2.cat.fastq.gz | Paired-end Illumina, Second of pair, FASTQ | Yeast ChIP-seq |
| human_rnaseq.fastq.gz | Paired-end Illumina, First of pair only, FASTQ | Human RNA-seq |
| human_mirnaseq.fastq.gz | Single-end Illumina, FASTQ | Human microRNA-seq |
| cholera_rnaseq.fastq.gz | Single-end Illumina, FASTQ | V. cholerae RNA-seq |
Reference Genomes
...
Here are the four reference genomes we will be using today, with some information about them. These are not necessarily the most recent versions of these references (e.g. the newest human reference genome is hg38 and the most a recent miRBase annotation is version is v21. (See here for information about many more genomes.)
...
We've discovered a pattern (also known as a regular expression) to use in searching, and the command line tool that does regular expression matching is grep (general regular expression parser). (Read more about grep here: Advanced commands: grep.and regular expressions)
Regular expressions are so powerful that nearly every modern computer language includes a "regex" module of some sort. There are many online tutorials for regular expressions, and several slightly different "flavors" of them. But the most common is the Perl style (http://perldoc.perl.org/perlretut.html), which was one of the fist and still the most powerful (there's a reason Perl was used extensively when assembling the human genome). We're only going to use simple regular expressions here, but learning more about them will pay handsome dividends for you in the future.
...
| Code Block |
|---|
| language | bash |
|---|
| title | grep to match contig names in a FASTA file |
|---|
|
# If you haven't stagedStage the fastaFASTA files
cds
mkdir -p core_ngs/references/fasta
cd core_ngs/references/fasta
cp $CORENGS/references/fasta/*.fa .
cd $SCRATCH/core_ngs/references/fasta
grep -P '^>' sacCer3.fa | more |
...
Now down to the nuts and bolts of the pattern: '^>'
First, the single quotes around the pattern – this tells the bash shell to pass the exact string contents to grep.
As part of its friendly we have seen, during command line parsing and evaluation , the shell will often look for special characters metacharacters on the command line that mean something to it (for example, the the $ in front of an environment variable name, like in $SCRATCH). Well, regular expressions treat the $ specially too – but in a completely different way! Those Those single quotes tell tell the shell "don't look inside here for special characters – treat this as a literal string and pass it to the program". The shell will obey, will strip the single quotes off the string, and will pass the actual pattern, ^>, to the grep program. (Note that the shell does look inside double quotes ( " ) for certain special signals, such as looking for environment variable names to evaluate. Read more about program. (Read more about Quoting in the shell.)
So what does ^> mean to grep? We know that contig name lines always start with a > character, so > is a literal for grep to use in its pattern match.
We might be able to get away with just using this literal alone as our regex, specifying '>' as as the command line pattern argument. But for for grep, the more specific the pattern, the better. So we constrain where the > can appear on the line. The special carat ( ^ ) metacharacter represents "beginning of line". So ^> means "beginning of a line followed by a > character".
Exercise: How many contigs are there in the sacCer3 reference?
| Expand |
|---|
|
| Code Block |
|---|
| language | bash |
|---|
| title | Get the alignment exercises files |
|---|
| # Copy the FASTA files for building references
mkdir -p $SCRATCH/core_ngs/references/fasta
cp $CORENGS/references/fasta/*.fa $SCRATCH/core_ngs/references/fasta/ |
|
Exercise: How many contigs are there in the sacCer3 reference?
| Expand |
|---|
|
| Code Block |
|---|
| cd $SCRATCH/core_ngs/references/fasta
grep -P '^>' sacCer3.fa | wc -l |
Or use grep's -c option that says "just count the line matches" | Code Block |
|---|
| grep -P -c '^>' sacCer3.fa |
|
...
- Trim the FASTQ sequences down to 50 with fastx_clipper
- this removes most of any 5' adapter contamination without the fuss of specific adapter trimming w/cutadapt
- Prepare the sacCer3 reference index for bwa using bwa index
- this is done once, and re-used for later alignments
- Perform a global bwa alignment on the R1 reads (bwa aln) producing a BWA-specific binary .sai intermediate file
- Perform a global bwa alignment on the R2 reads (bwa aln) producing a BWA-specific binary .sai intermediate file
- Perform pairing of the separately aligned reads and report the alignments in SAM format using bwa sampe
- Convert the SAM file to a BAM file (samtools view)
- Sort the BAM file by genomic location (samtools sort)
- Index the BAM file (samtools index)
- Gather simple alignment statistics (samtools flagstat and samtools idxstatidxstats)
We're going to skip the trimming step for now and see how it goes. We'll perform steps 2 - 5 now and leave samtools for a later exercise since steps 6 - 10 are common to nearly all post-alignment workflows.
...
Like other tools you've worked with so far, you first need to load bwa. Do that now, and then enter bwa with no arguments to view the top-level help page (many NGS tools will provide some help when called with no arguments). bwa is available as a BioContainers. module.
...
Make sure you're in
...
an idev session
| Code Block |
|---|
| language | bash |
|---|
| title | Start an idev session |
|---|
|
idev -m 120180 -N 1 -A OTH21164 -r CoreNGSday4CoreNGS-Thu
# or
idev -m 90 -N 1 -A OTH21164 -p development |
| Code Block |
|---|
|
module |
| Code Block |
|---|
|
module load biocontainers # takes a while
module load bwa
bwa |
...
| Expand |
|---|
|
| Code Block |
|---|
| language | bash |
|---|
| title | Get the alignment exercises files |
|---|
| mkdir -p $SCRATCH/core_ngs/alignment/fastq
mkdir -p $SCRATCH/core_ngs/references/fasta
cp $CORENGS/alignment/*fastq.gz $SCRATCH/core_ngs/alignment/fastq/
cp $CORENGS/references/fasta/*.fa $SCRATCH/core_ngs/references/fasta/ |
|
...
| Code Block |
|---|
| language | bash |
|---|
| title | Prepare BWA reference directory for sacCer3 |
|---|
|
mkdir -p $SCRATCH/core_ngs/references/bwa/sacCer3
cd $SCRATCH/core_ngs/references/bwa/sacCer3
ln -ssf ../../fasta/sacCer3.fa
ls -l |
...
| Expand |
|---|
|
| Code Block |
|---|
| # Copy the FASTA files for building references
mkdir -p $SCRATCH/core_ngs/references
cp $CORENGS/references/fasta/*.fa $SCRATCH/core_ngs/references/fasta/
# Copy a pre-built references bwa index for sacCer3
mkdir -p $SCRATCH/core_ngs/references/bwa/sacCer3
cp $CORENGS/references/bwa/sacCer3/*.fa* $SCRATCH/core_ngs/references/bwa/fastasacCer3/
# Get the FASTQ to align
mkdir -p $SCRATCH/core_ngs/alignment/fastq
cp $CORENGS/alignment/*fastq.gz $SCRATCH/core_ngs/alignment/fastq/
|
|
...
| Code Block |
|---|
| language | bash |
|---|
| title | Prepare to align yeast data |
|---|
|
mkdir -p $SCRATCH/core_ngs/alignment/yeast_bwa
cd $SCRATCH/core_ngs/alignment/yeast_bwa
ln -s -f .sf ../fastq
ln -s -fsf ../../references/bwa/sacCer3 |
...
Required arguments are a <prefix> of the bwa index files, and the input FASTQ file. There are lots of options, but here is a summary of the most important ones.
...
| Code Block |
|---|
| language | bash |
|---|
| title | bwa aln commands for yeast R1 and R2 |
|---|
|
# If not already loaded:
module load biocontainers
module load bwa
cd $SCRATCH/core_ngs/alignment/yeast_bwa
bwa aln sacCer3/sacCer3.fa fastq/Sample_Yeast_L005_R1.cat.fastq.gz > yeast_pe_R1.sai
bwa aln sacCer3/sacCer3.fa fastq/Sample_Yeast_L005_R2.cat.fastq.gz > yeast_pe_R2.sai |
When all is done you should have two .sai files: yeast_pe_R1.sai and yeast_pe_R2.sai.
| Tip |
|---|
| title | Make sure your output files are not empty |
|---|
|
Double check that output was written by doing ls -lh and making sure the file sizes listed are not 0. |
...
| Code Block |
|---|
bwa aln -t 60 sacCer3/sacCer3.fa fastq/Sample_Yeast_L005_R2.cat.fastq.gz > yeast_pe_R2.sai |
Exercise: How much of a speedup did you seen when aligning the R2 file with 20 60 threads?
| Expand |
|---|
|
The last few lines of bwa's execution output should look something like this: | Code Block |
|---|
| [bwa_aln] 17bp reads: max_diff = 2
[bwa_aln] 38bp reads: max_diff = 3
[bwa_aln] 64bp reads: max_diff = 4
[bwa_aln] 93bp reads: max_diff = 5
[bwa_aln] 124bp reads: max_diff = 6
[bwa_aln] 157bp reads: max_diff = 7
[bwa_aln] 190bp reads: max_diff = 8
[bwa_aln] 225bp reads: max_diff = 9
[bwa_aln_core] calculate SA coordinate... 266.70 sec
[bwa_aln_core] write to the disk... 0.04 sec
[bwa_aln_core] 262144 sequences have been processed.
[bwa_aln_core] calculate SA coordinate... 268.94 sec
[bwa_aln_core] write to the disk... 0.03 sec
[bwa_aln_core] 524288 sequences have been processed.
[bwa_aln_core] calculate SA coordinate... 72.26 sec
[bwa_aln_core] write to the disk... 0.01 sec
[bwa_aln_core] 592180 sequences have been processed.
[main] Version: 0.7.17-r1188
[main] CMD: /usr/local/bin/bwa aln -t 60 sacCer3/sacCer3.fa fastq/Sample_Yeast_L005_R2.cat.fastq.gz
[main] Real time: 5.013 sec; CPU: 142.813 sec |
So the R2 alignment took only ~5 seconds (real time), or 15+ times as fast as with only one processing thread. Note, though, that the CPU time with 60 threads was greater (142.8 sec) than with only 1 thread (77.6 sec). That's because of the thread management overhead when using multiple threads. |
...
Here is the command line statement you need. Just execute it on the command line.
| Expand |
|---|
|
|
| title | Pairing of BWA R1 and R2 aligned reads |
|---|
bwa sampe sacCer3/sacCer3.fa yeast_R1.sai yeast_R2.sai \
fastq/Sample_Yeast_L005_R1.cat.fastq.gz \
fastq/Sample_Yeast_L005_R2.cat.fastq.gz > yeast_pairedend.sam |
# Copy the FASTA files for building references
mkdir -p $SCRATCH/core_ngs/references
cp $CORENGS/references/fasta/*.fa $SCRATCH/core_ngs/references/fasta/
# Copy a pre-built bwa index for sacCer3
mkdir -p $SCRATCH/core_ngs/references/bwa/sacCer3
cp $CORENGS/references/bwa/sacCer3/*.* $SCRATCH/core_ngs/references/bwa/sacCer3/
# Get the FASTQ to align
mkdir -p $SCRATCH/core_ngs/alignment/fastq
cp $CORENGS/alignment/*fastq.gz $SCRATCH/core_ngs/alignment/fastq/
# Stage the BWA .sai files
mkdir -p $SCRATCH/core_ngs/alignment/yeast_bwa
cd $SCRATCH/core_ngs/alignment/yeast_bwa
ln -sf ../fastq
ln -sf ../../references/bwa/sacCer3
cp $CORENGS/catchup/yeast_bwa/*.sai .
|
|
| Code Block |
|---|
| language | bash |
|---|
| title | Pairing of BWA R1 and R2 aligned reads |
|---|
|
cd $SCRATCH/core_ngs/alignment/yeast_bwa
bwa sampe sacCer3/sacCer3.fa yeast_pe_R1.sai yeast_pe_R2.sai \
fastq/Sample_Yeast_L005_R1.cat.fastq.gz \
fastq/Sample_Yeast_L005_R2.cat.fastq.gz > yeast_pe.sam |
You should now have a SAM file (yeast_pe.sam) that contains the alignments.
Exercise: How many lines does the SAM file have? How does this compare to the number of input sequences (R1+R2)?
| Expand |
|---|
|
wc -l yeast_pe.sam reports 1,184,378 lines The alignment SAM file will contain records for both R1 and R2 reads, so we need to count sequences in both files. zcat ./fastq/Sample_Yeast_L005_R[12]*gz | wc -l | awk '{print $1/4}' reports 1,184,360 reads that were aligned So the SAM file has 18 more lines than the R1+R2 total. These are the header records that appear before any alignment records. |
You should now have a SAM file (yeast_pairedend.sam) that contains the alignments. It's just a text file, so take a look with head, more, less, tail, or whatever you feel like. Later you'll learn additional ways to analyze the data with samtools once you create a BAM file.
...
| Expand |
|---|
|
This looks for the pattern '^HWI' which is the start of every read name (which starts every alignment record).
Remember -c says just count the records, don't display them. | Code Block |
|---|
| grep -P -c '^HWI' yeast_pairedendpe.sam |
Or use the -v (invert) option to tell grep to print all lines that don't match a particular pattern; here, all header lines, which start with @. | Code Block |
|---|
| grep -P -v -c '^@' yeast_pairedendpe.sam |
|
| Expand |
|---|
|
| There are 1,184,360 alignment records. |
Exercise: How many sequences were in the R1 and R2 FASTQ files combined?
| Expand |
|---|
|
zcat fastq/Sample_Yeast_L005_R[12].cat.fastq.gz | wc -l | awk '{print $1/4}'
|
| Expand |
|---|
|
| There were a total of 1,184,360 original sequences (R1s + R2s) |
Exercises:
...
Exercises:
- Does the SAM file contain both mapped and un-mapped reads?
- What is the order of the alignment records in this SAM file?
| Expand |
|---|
|
Both R1 and R2 reads must have separate alignment records, because there were 1,184,360 R1+R2 reads and the same number of alignment records. The SAM file must contain both mapped and un-mapped reads, because there were 1,184,360 R1+R2 reads and the and the same number of alignment records. Alignment records occur in the same read-name order as they did in the FASTQ, except that they come in pairs. The R1 read comes 1st, then the corresponding R2. This is called read name ordering. |
...
Suppose you wanted to look only at field 3 (contig name) values in the SAM file. You can do this with the handy cut command. Below is a simple example where you're asking cut to display the 3rd column value for the last 10 alignment records.
| Expand |
|---|
|
| Code Block |
|---|
| # Stage the aligned SAM file
mkdir -p $SCRATCH/core_ngs/alignment/yeast_bwa
cd $SCRATCH/core_ngs/alignment/yeast_bwa
cp $CORENGS/catchup/yeast_bwa/yeast_pe.sam .
|
|
| Code Block |
|---|
| language | bash |
|---|
| title | Cut syntax for a single field |
|---|
|
tail yeast_pairedendpe.sam | cut -f 3 |
By default cut assumes the field delimiter is Tab, which is the delimiter used in the majority of NGS file formats. You can specify a different delimiter with the -d option.
...
| Code Block |
|---|
| language | bash |
|---|
| title | Cut syntax for multiple fields |
|---|
|
tail -20 yeast_pairedendpe.sam | cut -f 2-6,9 |
You may have noticed that some alignment records contain contig names (e.g. chrV) in field 3 while others contain an asterisk ( * ). The * means the record didn't map. We're going to use this heuristic along with cut to see about how many records represent aligned sequences. (Note this is not the strictly correct method of finding unmapped reads because not all unmapped reads have an asterisk in field 3. Later you'll see how to properly distinguish between mapped and unmapped reads using samtools.)
...
| Code Block |
|---|
| language | bash |
|---|
| title | Grep pattern that doesn't match header |
|---|
|
# the ^@ pattern matches lines starting with @ (only header lines),
# and -v says output lines that don't match
grep -v -P '^@' yeast_pairedendpe.sam | head |
Ok, it looks like we're seeing only alignment records. Now let's pull out only field 3 using cut:
...
| Code Block |
|---|
| language | bash |
|---|
| title | Filter contig name of * (unaligned) |
|---|
|
grep -v -P '^@' yeast_pairedendpe.sam | cut -f 3 | grep -v '*' | head |
...
| Code Block |
|---|
| language | bash |
|---|
| title | Count aligned SAM records |
|---|
|
grep -v -P '^@' yeast_pairedendpe.sam | cut -f 3 | grep -v '*' | wc -l |
...
| Expand |
|---|
| title | Make sure you're in a idev session |
|---|
|
| Code Block |
|---|
| language | bash |
|---|
| title | Start an idev session |
|---|
| idev -m 120 -N 1 -A OTH21164 -r CoreNGSday4 CoreNGS-Thu
# or
idev -m 90 -N 1 -A OTH21164 -p development |
|
| Code Block |
|---|
|
# If not already loaded
module load biocontainers # takes a while
module load samtools
samtools |
...
In this exercise, we will explore five utilities provided by samtools: view, sort, index, flagstat, and idxstats. Each of these is executed in one line for a given SAM/BAM file. In the SAMtools/BEDtools sections tomorrow we will explore samtools in capabilities more in depth.
| Warning |
|---|
| title | Know your samtools version! |
|---|
|
There are two main "eras" of SAMtools development: - "original" samtools
- v 0.1.19 is the last stable version
- "modern" samtools
- v 1.0, 1.1, 1.2 – avoid these (very buggy!)
- v 1.3+ – finally stable!
Unfortunately, some functions with the same name in both version eras have different options and arguments! So be sure you know which version you're using. (The samtools version is usually reported at the top of its usage listing). TACC BioContainers also offers the original samtools version: samtools/ctr-0.1.19--3. |
...
| Expand |
|---|
|
| Code Block |
|---|
| language | bash |
|---|
| title | Get the alignment exercises files |
|---|
| mkdir -p $SCRATCH/core_ngs/alignment/yeast_bwa
cd $SCRATCH/core_ngs/alignment/yeast_bwa
cp $CORENGS/catchup/yeast_bwa/yeast_pairedendpe.sam .
|
|
| Code Block |
|---|
| language | bash |
|---|
| title | Convert SAM to binary BAM |
|---|
|
cd $SCRATCH/core_ngs/alignment/yeast_bwa
cat yeast_pairedend.sam | samtools view -b -oyeast_pe.sam > yeast_pairedendpe.bam |
- the -b option tells the tool to output BAM format
- the -o option specifies the name of the output BAM file that will be created
- we pipe the entire SAM file to samtools view so that the header records are included (required for SAM → BAM conversion)
- samtools view reads its input from standard input by default
- the tool to output BAM format
The BAM file is a binary file, not a text file, so how do you look at its contents now? How do you look at the BAM file contents now? That's simple. Just use samtools view without the -b option. Remember to pipe output to a pager!
| Code Block |
|---|
| language | bash |
|---|
| title | View BAM records |
|---|
|
samtools view yeast_pairedendpe.bam | more
|
Notice that this does not show us the header record we saw at the start of the SAM file.
Exercise: What samtools view option will include the header records in its output? Which option would show only the header records?
| Expand |
|---|
|
samtools view | less
then search for "header" ( /header ) |
| Expand |
|---|
|
samtools view -h shows header records along with alignment records. samtools view -H shows header records only. |
...
Looking at some of the alignment record information (e.g. samtools view yeast_pairedendpe.bam | cut -f 1-4 | more), you will notice that read names appear in adjacent pairs (for the R1 and R2), in the same order they appeared in the original FASTQ file. Since that means the corresponding mappings are in no particular order, searching through the file very inefficient. samtools sort re-orders entries in the SAM file either by locus (contig name + coordinate position) or by read name.
...
| Expand |
|---|
|
Copy aligned yeast BAM file | Code Block |
|---|
| # Stage the aligned yeast SAM and BAM files
mkdir -p $SCRATCH/core_ngs/alignment/yeast_bwa
cd $SCRATCH/core_ngs/alignment/yeast_bwa
cp $CORENGS/catchup/yeast_bwa/yeast_pairedend.bampe.[bs]am . |
|
To sort the paired-end yeast BAM file by position, and get a BAM file named yeast_pairedendpe.sort.bam as output, execute the following command:
| Code Block |
|---|
| language | bash |
|---|
| title | Sort a BAM file |
|---|
|
cd $SCRATCH/core_ngs/alignment/yeast_bwa
samtools sort -O bam -T yeast_pairedendpe.tmp yeast_pairedendpe.bam > yeast_pairedendpe.sort.bam |
- The -O options says the Output format should be BAM
- The -T options gives a prefix for Temporary files produced during sorting
- sorting large BAMs will produce many temporary files during processingprocessing
- make sure the temporary file prefix is different from the input BAM file prefix!
- By default sort writes its output to standard output, so we use > to redirect to a file named yeast_pairedend.sort.bam
Exercise: Compare the file sizes of the yeast_pariedend pe .sam, .bam, and .sort.bam files and explain why they are different.
| Expand |
|---|
|
| Code Block |
|---|
| ls -lh yeast_pairedendpe.* |
|
| Expand |
|---|
|
The yeast_pairedendpe.sam text file is the largest at ~348 MB because it is an uncompressed text file. The name-ordered binary yeast_pairedendpe.bam text file only about 1/3 that size, ~111 MB. They contain exactly the same records, in the same order, but conversion from text to binary results in a much smaller file. The coordinate-ordered binary yeast_pairedendpe.sort.bam file is even slightly smaller, ~92 MB. This is because BAM files are actually customized gzip-format files. The customization allows blocks of data (e.g. all alignment records for a contig) to be represented in an even more compact form. You can read more about this in section 4 of the SAM format specification. |
...
| Code Block |
|---|
| language | bash |
|---|
| title | Index a sorted bam |
|---|
|
samtools index yeast_pairedendpe.sort.bam |
This will produce a file named yeast_pairedendpe.bam.bai.
Most of the time when an index is required, it will be automatically located as long as it is in the same directory as its BAM file and shares the same name up until the .bai extension.
...
| Expand |
|---|
|
| Code Block |
|---|
| ls -lh yeast_pairedendpe.sort.bam* |
|
| Expand |
|---|
|
While the yeast_pairedendpe.sort.bam file is ~92 MB, its index (yeast_pairedendpe.sort.bai) is only 20 KB. |
samtools flagstat
...
Here's how to run samtools flagstat and both see the output in the terminal and save it in a file – the samtools flagstat standard output is piped to tee, which both writes it to the specified file and sends it to its standard output:to the specified file and sends it to its standard output:
| Expand |
|---|
|
| Code Block |
|---|
| # Stage the aligned yeast SAM and BAM files
mkdir -p $SCRATCH/core_ngs/alignment/yeast_bwa
cd $SCRATCH/core_ngs/alignment/yeast_bwa
cp $CORENGS/catchup/yeast_bwa/yeast_pe.sort.bam* . |
|
| Code Block |
|---|
| language | bash |
|---|
| title | Run samtools flagstat using tee |
|---|
|
samtools flagstat yeast_pairedendpe.sort.bam | tee yeast_pariedendpe.flagstat.txt |
You should see something like this:
...
More information about the alignment is provided by the samtools idxstats report, which shows how many reads aligned to each contig in your reference. Note that samtools idxstats must be run on a sorted, indexed BAM file.
| Expand |
|---|
|
| Code Block |
|---|
| # Stage the aligned yeast SAM and BAM files
mkdir -p $SCRATCH/core_ngs/alignment/yeast_bwa
cd $SCRATCH/core_ngs/alignment/yeast_bwa
cp $CORENGS/catchup/yeast_bwa/yeast_pe.sort.bam* . |
|
| Code Block |
|---|
| language | bash |
|---|
| title | Use samtools idxstats to summarize mapped reads by contig |
|---|
|
samtools idxstats yeast_pairedendpe.sort.bam | tee yeast_pairedendpe.idxstats.txt |
Here we use the tee command which reports its standard input to standard output before also writing it to the specified file.
...