| Table of Contents |
|---|
Overview
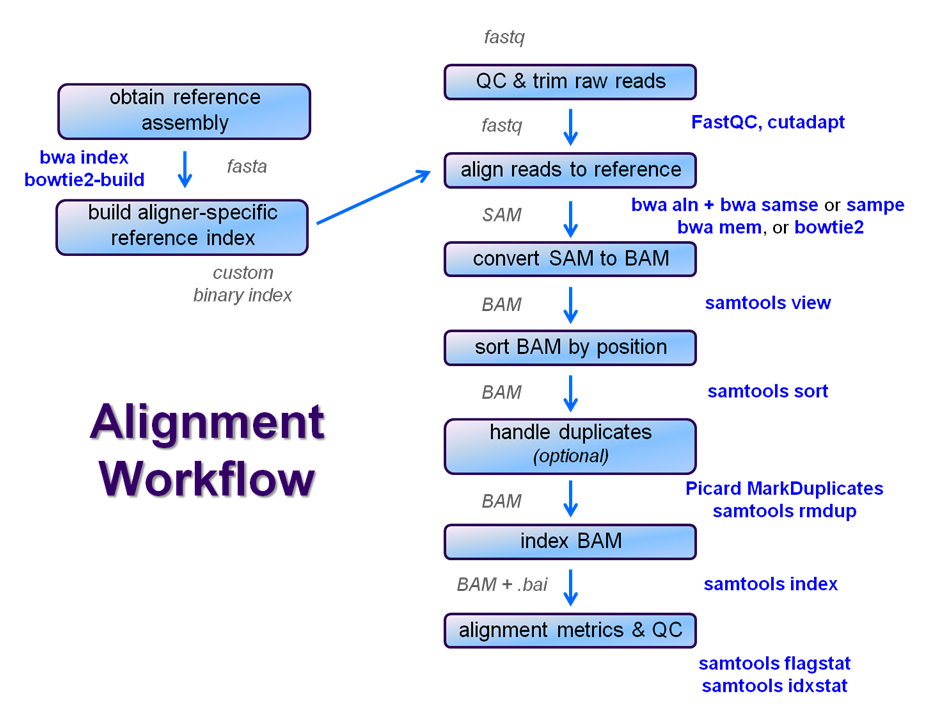
After raw sequence files are generated (in FASTQ format), quality-checked, and pre-processed in some way, the next step in many NGS pipelines is mapping to a reference genome.
...
Even though many mapping tools exist, a few individual programs have a dominant "market share" of the NGS world. In this section, we will primarily focus on two of the most versatile general-purpose ones: BWA and Bowtie2 (the latter being part of the Tuxedo suite which includes the transcriptome-aware RNA-seq aligner Tophat2 as well as other downstream quantifiaction tools).
Stage the alignment data
First connect to stampede2.tacc.utexas.edu and start an idev session. This should be second nature by now ![]()
| Code Block | ||||
|---|---|---|---|---|
| ||||
idev -p developmentnormal -m 120180 -A UT-2015-05-18 -N 1 -n 68 --reservation=BIO_DATA_week_1 |
Then stage the sample datasets and references we will use.
| Code Block | ||||
|---|---|---|---|---|
| ||||
mkdir -p $SCRATCH/core_ngs/references/fasta
mkdir -p $SCRATCH/core_ngs/alignment/fastq
cp $CORENGS/references/*.fa $SCRATCH/core_ngs/references/fasta/
cp $CORENGS/alignment/*fastq.gz $SCRATCH/core_ngs/alignment/fastq/
cd $SCRATCH/core_ngs/alignment/fastq |
...
| File Name | Description | Sample |
|---|---|---|
| Sample_Yeast_L005_R1.cat.fastq.gz | Paired-end Illumina, First of pair, FASTQ | Yeast ChIP-seq |
| Sample_Yeast_L005_R2.cat.fastq.gz | Paired-end Illumina, Second of pair, FASTQ | Yeast ChIP-seq |
| human_rnaseq.fastq.gz | Paired-end Illumina, First of pair only, FASTQ | Human RNA-seq |
| human_mirnaseq.fastq.gz | Single-end Illumina, FASTQ | Human microRNA-seq |
| cholera_rnaseq.fastq.gz | Single-end Illumina, FASTQ | V. cholerae RNA-seq |
Reference Genomes
Before we get to alignment, we need a reference to align to. This is usually an organism's genome, but can also be any set of names sequences, such as a transcriptome or other set of genes.
...
| Tip | |||||
|---|---|---|---|---|---|
The BioITeam maintains a set of reference indexes for many common organisms and aligners. They can be found in aligner-specific sub-directories of the /work2/projects/BioITeam/ref_genome area. E.g.:
|
Exploring FASTA with grep
It is often useful to know what chromosomes/contigs are in a FASTA file before you start an alignment so that you're familiar with the contig naming convention – and to verify that it's the one you expect. For example, chromosome 1 is specified differently in different references and organisms: chr1 (USCS human), chrI (UCSC yeast), or just 1 (Ensembl human GRCh37).
...
| Expand | ||
|---|---|---|
| ||
There are 17 contigs. |
Aligner overview
There are many aligners available, but we will concentrate on two of the most popular general-purpose ones: bwa and bowtie2. The table below outlines the available protocols for them.
| alignment type | aligner options | pro's | con's |
|---|---|---|---|
| global with bwa | SE:
PE:
|
|
|
| global with bowtie2 | bowtie2 --global |
|
|
| local with bwa | bwa mem |
|
|
| local with bowtie2 | bowtie2 --local |
|
|
Exercise #1: BWA global alignment – Yeast ChIP-seq
Overview ChIP-seq alignment workflow with BWA
We will perform a global alignment of the paired-end Yeast ChIP-seq sequences using bwa. This workflow has the following steps:
...
We're going to skip the trimming step for now and see how it goes. We'll perform steps 2 - 5 now and leave samtools for a later exercise since steps 6 - 10 are common to nearly all post-alignment workflows.
Introducing BWA
Like other tools you've worked with so far, you first need to load bwa. Do that now, and then enter bwa with no arguments to view the top-level help page (many NGS tools will provide some help when called with no arguments). Note that bwa is available both from the standard TACC module system and as BioContainers. module.
| Expand | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| an idev session. If you're in an idev session, the hostname command will display a name like c455-021.stampede2.tacc.utexas.edu. But if you're on a login node the hostname will be something like login3.stampede2.tacc.utexas.edu.If you're on
| login node, start an
| like this:|||||||
|
| Code Block | ||
|---|---|---|
| ||
module load biocontainers # takes a while module load bwa bwa |
...
As you can see, bwa include many sub-commands that perform the tasks we are interested in.
Building the BWA sacCer3 index
We will index the genome with the bwa index command. Type bwa index with no arguments to see usage for this sub-command.
...
| Code Block | ||
|---|---|---|
| ||
sacCer3.fa sacCer3.fa.amb sacCer3.fa.ann sacCer3.fa.bwt sacCer3.fa.pac sacCer3.fa.sa |
Performing the bwa alignment
Now, we're ready to execute the actual alignment, with the goal of initially producing a SAM file from the input FASTQ files and reference. First prepare a directory for this exercise and link the sacCer3 reference directories there (this will make our commands more readable).
...
| Expand | ||
|---|---|---|
| ||
Both R1 and R2 reads must have separate alignment records, because there were 1,184,360 R1+R2 reads and the same number of alignment records. The SAM file must contain both mapped and un-mapped reads, because there were 1,184,360 R1+R2 reads and the same number of alignment records. Alignment records occur in the same read-name order as they did in the FASTQ, except that they come in pairs. The R1 read comes 1st, then the corresponding R2. This is called read name ordering. |
Using cut to isolate fields
Recall the format of a SAM alignment record:
...
First we need to make sure that we don't look at fields in the SAM header lines. We're going to end up with a series of pipe operations, and the best way to make sure you're on track is to enter them one at a time piping to head:
...
| Expand | |||||||
|---|---|---|---|---|---|---|---|
| |||||||
The expression above returns 612,968. There were 1,184,360 records total, so the percentage is:
or about 51%. Not great. Note we perform this calculation in awk's BEGIN block, which is always executed, instead of the body block, which is only executed for lines of input. And here we call awk without piping it any input. See Linux fundamentals: cut,sort,uniq,grep,awk |
Exercise: What might we Exercise: What might we try in order to improve the alignment rate?
| Expand | ||
|---|---|---|
| ||
| Recall that these are 100 bp reads and we did not remove adapter contamination. There will be a distribution of fragment sizes – some will be short – and those short fragments may not align without adapter removal (e.g. with fastx_trimmer). |
Exercise #2: Basic SAMtools Utilities
The SAMtools program is a commonly used set of tools that allow a user to manipulate SAM/BAM files in many different ways, ranging from simple tasks (like SAM/BAM format conversion) to more complex functions (like sorting, indexing and statistics gathering). It is available in the TACC module system (as well as in BioContainers). Load that module and see what samtools has to offer:
| Expand | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| you're in an idev session. If
| an idev session, the hostname command will display a name like c455-021.stampede2.tacc.utexas.edu. But if you're on a login node the hostname will be something like login3.stampede2.tacc.utexas.edu.If you're on
| login node, start an
| like this:||||||||
|
| Code Block | ||
|---|---|---|
| ||
# If not already loaded module load biocontainers # takes a while module load samtools samtools |
...
| Warning | ||
|---|---|---|
| ||
There are two main "eras" of SAMtools development:
Unfortunately, some functions with the same name in both version eras have different options and arguments! So be sure you know which version you're using. (The samtools version is usually reported at the top of its usage listing). TACC BioContainers also offers the original samtools version: samtools/ctr-0.1.19--3. |
samtools view
The samtools view utility provides a way of converting between SAM (text) and BAM (binary, compressed) format. It also provides many, many other functions which we will discuss lster. To get a preview, execute samtools view without any other arguments. You should see:
...
| Expand | ||
|---|---|---|
| ||
samtools view -h shows header records along with alignment records. samtools view -H shows header records only. |
samtools sort
Looking at some of the alignment record information (e.g. samtools view yeast_pairedend.bam | cut -f 1-4 | more), you will notice that read names appear in adjacent pairs (for the R1 and R2), in the same order they appeared in the original FASTQ file. Since that means the corresponding mappings are in no particular order, searching through the file very inefficient. samtools sort re-orders entries in the SAM file either by locus (contig name + coordinate position) or by read name.
...
| Expand | ||
|---|---|---|
| ||
The yeast_pairedend.sam text file is the largest at ~348 MB. The name-ordered binary yeast_pairedend.bam text file only about 1/3 that size, ~110 MB. They contain exactly the same records, in the same order, but conversion from text to binary results in a much smaller file. The coordinate-ordered binary yeast_pairedend.sort.bam file is even slightly smaller, ~91 MB. This is because BAM files are actually customized gzip-format files. The customization allows blocks of data (e.g. all alignment records for a contig) to be represented in an even more compact form. You can read more about this in section 4 of the SAM format specification. |
samtools index
Many tools (like IGV, the Integrative Genomics Viewer) only need to use portions of a BAM file at a given point in time. For example, if you are viewing alignments that are within a particular gene, alignment records on other chromosomes do not need to be loaded. In order to speed up access, BAM files are indexed, producing BAI files which allow fast random access. This is especially important when you have many alignment records.
...
| Expand | ||
|---|---|---|
| ||
While the yeast_pairedend.sort.bam text file is ~91 MB, its index (yeast_pairedend.sort.bai) is only 20 KB. |
samtools flagstat
Since the BAM file contains records for both mapped and unmapped reads, just counting records doesn't provide information about the mapping rate of our alignment. The samtools flagstat tool provides a simple analysis of mapping rate based on the the SAM flag fields.
...
| Expand | ||
|---|---|---|
| ||
About 86% of mapped read were properly paired. This is actually a bit on the low side for ChIP-seq alignments which typically over 90%. |
samtools idxstats
More information about the alignment is provided by the samtools idxstats report, which shows how many reads aligned to each contig in your reference. Note that samtools idxstats must be run on a sorted, indexed BAM file.
...
| Tip |
|---|
If you're mapping to a non-genomic reference such as miRBase miRNAs or another set of genes (a transcriptome), samtools idxstats gives you a quick look at quantitative alignment results. |
Exercise #3: PE alignment with BioITeam scripts
Now that you've done everything the hard way, let's see how to do run an alignment pipeline using a BWA alignment script maintained by the BioITeam, /work2/projects/BioITeam/common/script/align_bwa_illumina.sh. Type in the script name to see its usage.
| Code Block | ||
|---|---|---|
| ||
align_bwa_illumina.sh 2021_06_05
Align Illumina SE or PE data with bwa. Produces a sorted, indexed,
duplicate-marked BAM file and various statistics files. Usage:
align_bwa_illumina.sh <aln_mode> <in_file> <out_pfx> <assembly> [ paired trim_sz trim_sz2 seq_fmt qual_fmt ]
Required arguments:
aln_mode Alignment mode, either global (bwa aln) or local (bwa mem).
in_file For single-end alignments, path to input sequence file.
For paired-end alignments using fastq, path to the the R1
fastq file which must contain the string 'R1' in its name.
The corresponding 'R2' must have the same path except for 'R1'.
out_pfx Desired prefix of output files in the current directory.
assembly One of hg38, hg19, hg38, mm10, mm9, sacCer3, sacCer1, ce11, ce10,
danRer7, hs_mirbase, mm_mirbase, or reference index prefix.
Optional arguments:
paired 0 = single end alignment (default); 1 = paired end.
trim_sz Size to trim reads to. Default 0 (no trimming)
trim_sz2 Size to trim R2 reads to for paired end alignments.
Defaults to trim_sz
seq_fmt Format of sequence file (fastq, bam or scarf). Default is
fastq if the input file has a '.fastq' extension; scarf
if it has a '.sequence.txt' extension.
qual_type Type of read quality scores (sanger, illumina or solexa).
Default is sanger for fastq, illumina for scarf.
Environment variables:
show_only 1 = only show what would be done (default not set)
aln_args other bowtie2 options (e.g. '-T 20' for mem, '-l 20' for aln)
no_markdup 1 = don't mark duplicates (default 0, mark duplicates)
run_fastqc 1 = run fastqc (default 0, don't run). Note that output
will be in the directory containing the fastq files.
keep 1 = keep unsorted BAM (default 0, don't keep)
bwa_bin BWA binary to use. Default bwa 0.7.x. Note that bwa 0.6.2
or earlier should be used for scarf and other short reads.
also: NUM_THREADS, BAM_SORT_MEM, SORT_THREADS, JAVA_MEM_ARG
Examples:
align_bwa_illumina.sh local ABC_L001_R1.fastq.gz my_abc hg38 1
align_bwa_illumina.sh global ABC_L001_R1.fastq.gz my_abc hg38 1 50
align_bwa_illumina.sh global sequence.txt old sacCer3 0 '' '' scarf solexa |
There are lots of bells and whistles in the arguments, but the most important are the first few:
- aln_mode – whether to perform a global or local alignment (the 1st argument must be one of those words)
- global mode uses the bwa aln workflow as we did above
- local mode uses the bwa mem command
- in_file – full or relative path to the FASTQ file (just the R1 fastq if paired end). Can be compressed (.gz)
- out_pfx – prefix for all the output files produced by the script. Should relate back to what the data is.
- assembly – genome assembly to use.
- there are pre-built indexes for some common eukaryotes (hg38, hg19, mm10, mm9, danRer7, sacCer3) that you can use
- or provide a full path for a bwa reference index you have built somewhere
- paired flag – 0 means single end (the default); 1 means paired end
- trim_sz – if you want the FASTQ hard trimmed down to a specific length before alignment, supply that number here
We're going to run this script and a similar Bowtie2 alignment script, on the yeast data using the TACC batch system. In a new directory, copy over the commands and submit the batch job. We ask for 2 hours (-t 02:00:00) with 4 tasks/node (-w 4); since we have 4 commands, this will run on 1 compute node.
| Expand | |||||
|---|---|---|---|---|---|
| |||||
|
| Code Block | ||||
|---|---|---|---|---|
| ||||
# Make sure you're not in an idev session by looking at the hostname
hostname
# If the hostname looks like "c455-004.stampede2.tacc.utexas.edu", exit the idev session
# Make a new alignment directory for running these scripts
mkdir -p $SCRATCH/core_ngs/alignment/bwa_script
cd $SCRATCH/core_ngs/alignment/bwa_script
ln -s -f ../fastq
# Copy the alignment commands file and submit the batch job
cp $CORENGS/tacc/aln_script.cmds .
launcher_creator.py -j aln_script.cmds -n aln_script -t 02:00:00 -w 4 -a UT-2015-05-18 -q normal
sbatch --reservation=BIO_DATA_week_1 aln_script.slurm
showq -u |
While we're waiting for the job to complete, lets look at the aln_script.cmds file.
| Code Block | ||||
|---|---|---|---|---|
| ||||
/work2/projects/BioITeam/common/script/align_bwa_illumina.sh global ./fastq/Sample_Yeast_L005_R1.cat.fastq.gz bwa_global sacCer3 1 50
/work2/projects/BioITeam/common/script/align_bwa_illumina.sh local ./fastq/Sample_Yeast_L005_R1.cat.fastq.gz bwa_local sacCer3 1
/work2/projects/BioITeam/common/script/align_bowtie2_illumina.sh global ./fastq/Sample_Yeast_L005_R1.cat.fastq.gz bt2_global sacCer3 1 50
/work2/projects/BioITeam/common/script/align_bowtie2_illumina.sh local ./fastq/Sample_Yeast_L005_R1.cat.fastq.gz bt2_local sacCer3 1 |
Notes:
- The 1st command performs a paired-end BWA global alignment (similar to above), but asks that the 100 bp reads be trimmed to 50 first.
- we refer to the pre-built index for yeast by name: sacCer3
- this index is located in the /work/projects/BioITeam/ref_genome/bwa/bwtsw/sacCer3/ directory
- we provide the name of the R1 FASTQ file
- because we request a PE alignment (the 1 argument) the script will look for a similarly-named R2 file.
- all output files associated with this command will be named with the prefix bwa_global.
- we refer to the pre-built index for yeast by name: sacCer3
- The 2nd command performs a paired-end BWA local alignment.
- all output files associated with this command will be named with the prefix bwa_local.
- no trimming is requested because the local alignment should ignore 5' and 3' bases that don't match the reference genome
- The 3rd command performs a paired-end Bowtie2 global alignment.
- the Bowtie2 alignment script has the same first arguments as the BWA alignment script.
- all output files associated with this command will be named with the prefix bt2_global.
- again, we specify that reads should first be trimmed to 50 bp.
- The 4th command performs a paired-end Bowtie2 local alignment.
- all output files associated with this command will be named with the prefix bt2_local.
- again, no trimming is requested for the local alignment.
Output files
This alignment pipeline script performs the following steps:
- Hard trims FASTQ, if optionally specified (fastx_trimmer)
- Performs the global or local alignment (here, a PE alignment)
- BWA global: bwa aln the R1 and R2 separately, then bwa sampe to produce a SAM file
- BWA local: call bwa mem with both R1 and R2 to produce a SAM file
- Bowtie2 global: call bowtie2 --global with both R1 and R2 to produce a SAM file
- Bowtie2 local: call bowtie2 --local with both R1 and R2 to produce a SAM file
- BWA global: bwa aln the R1 and R2 separately, then bwa sampe to produce a SAM file
- Converts SAM to BAM (samtools view)
- Sorts the BAM (samtools sort)
- Marks duplicates (Picard MarkDuplicates)
- Indexes the sorted, duplicate-marked BAM (samtools index)
- Gathers statistics (samtools idxstats, samtools flagstat, plus a custom statistics script of Anna's)
- Removes intermediate files
There are a number of output files, with the most important being those desribed below.
- <prefix>.align.log – Log file of the entire alignment process.
- check the tail of this file to make sure the alignment was successful
- <prefix>.sort.dup.bam – Sorted, duplicate-marked alignment file.
- <prefix>.sort.dup.bam.bai – Index for the sorted, duplicate-marked alignment file
- <prefix>.flagstat.txt – samtools flagstat output
- <prefix>.idxstats.txt – samtools idxstats output
- <prefix>.samstats.txt – Summary alignment statistics from Anna's stats script
- <prefix>.iszinfo.txt – Insert size statistics (for paired-end alignments) from Anna's stats script
Verifying alignment success
The alignment log will have a "I ran successfully" message at the end if all went well, and if there was an error, the important information should also be at the end of the log file. So you can use tail to check the status of an alignment. For example:
| Code Block | ||||
|---|---|---|---|---|
| ||||
tail bwa_global.align.log |
This will show something like:
| Code Block |
|---|
..Done alignmentUtils.pl bamstats - 2020-06-14 23:19:38
.. samstats file 'bwa_global.samstats.txt' exists and is not empty - 2020-06-14 23:19:38
===============================================================================
## Cleaning up files (keep 0) - 2020-06-14 23:19:38
===============================================================================
ckRes 0 cleanup
===============================================================================
## All bwa alignment tasks completed successfully! - 2020-06-14 23:19:38
=============================================================================== |
Notice that success message: "All bwa alignment tasks completed successfully!". It should only appear once in any successful alignment log.
When multiple alignment commands are run in parallel it is important to check them all, and you can use grep looking for part of the unique success message to do this. For example:
| Code Block | ||||
|---|---|---|---|---|
| ||||
grep 'completed successfully!' *align.log | wc -l |
If this command returns 4 (the number of alignment tasks we performed), all went well, and we're done.
But what if something went wrong? How can we tell which alignment task was not successful? You could tail the log files one by one to see which one(s) don't have the message, but you can also use a special grep option to do this work.
| Code Block | ||||
|---|---|---|---|---|
| ||||
grep -L 'completed successfully' *.align.log |
The -L option tells grep to only print the filenames that don't contain the pattern. Perfect! To see happens in the case of failure, try it on a file that doesn't contain that message:
| Code Block | ||
|---|---|---|
| ||
grep -L 'completed successfully' aln_script.cmds |
Checking alignment statistics
The <prefix>.samstats.txt statistics files produced by the alignment pipeline has a lot of good information in one place. If you look at bwa_global.samstats.txt you'll see something like this:
| Code Block | ||
|---|---|---|
| ||
-----------------------------------------------
Aligner: bwa
Total sequences: 1184360
Total mapped: 539079 (45.5 %)
Total unmapped: 645281 (54.5 %)
Primary: 539079 (100.0 %)
Secondary:
Duplicates: 249655 (46.3 %)
Fwd strand: 267978 (49.7 %)
Rev strand: 271101 (50.3 %)
Unique hit: 503629 (93.4 %)
Multi hit: 35450 (6.6 %)
Soft clip:
All match: 531746 (98.6 %)
Indels: 7333 (1.4 %)
Spliced:
-----------------------------------------------
Total PE seqs: 1184360
PE seqs mapped: 539079 (45.5 %)
Num PE pairs: 592180
F5 1st end mapped: 372121 (62.8 %)
F3 2nd end mapped: 166958 (28.2 %)
PE pairs mapped: 80975 (13.7 %)
PE proper pairs: 16817 (2.8 %)
----------------------------------------------- |
Since this was a paired end alignment there is paired-end specific information reported.
You can also view statistics on insert sizes for properly paired reads in the bwa_global.iszinfo.txt file. This tells you the average (mean) insert size, standard deviation, mode (most common value), and fivenum values (minimum, 1st quartile, median, 3rd quartile, maximum).
| Code Block | ||
|---|---|---|
| ||
Insert size stats for: bwa_global
Number of pairs: 16807 (proper)
Number of insert sizes: 406
Mean [-/+ 1 SD]: 296 [176 416] (sd 120)
Mode [Fivenum]: 228 [51 224 232 241 500] |
A quick way to check alignment stats if you have run multiple alignments is again to use grep. For example:
| Code Block | ||||
|---|---|---|---|---|
| ||||
grep 'Total mapped' *samstats.txt |
will produce output like this:
| Code Block |
|---|
bt2_global.samstats.txt: Total mapped: 602893 (50.9 %)
bt2_local.samstats.txt: Total mapped: 788069 (66.5 %)
bwa_global.samstats.txt: Total mapped: 539079 (45.5 %)
bwa_local.samstats.txt: Total mapped: 1008000 (76.5 % |
Exercise: How would you list the median insert size for all the alignments?
| Expand | ||
|---|---|---|
| ||
That information is in the *.iszinfo.txt files, on the line labeled Mode. The median value is th 3rd value in the 5 fivnum values; it is the 7th whitespace-separated field on the Mode line. |
| Expand | |||||
|---|---|---|---|---|---|
| |||||
Use grep to isolate the Mode line, and awk to isolate the median value field:
|
TACC batch system considerations
The great thing about pipeline scripts like this is that you can perform alignments on many datasets in parallel at TACC, and they are written to take advantage of having multiple cores on TACC nodes where possible.
On the stampede2, with its 68 physical cores per node, they are designed to run best with no more than 4 tasks per node.
| Tip | ||
|---|---|---|
| ||
These alignment scripts should always be run with a wayness of 4 (-w 4) in the stampede2 batch system, meaning at most 4 commands per node. |