Table of Contents
Overview
...
Overview and Objectives
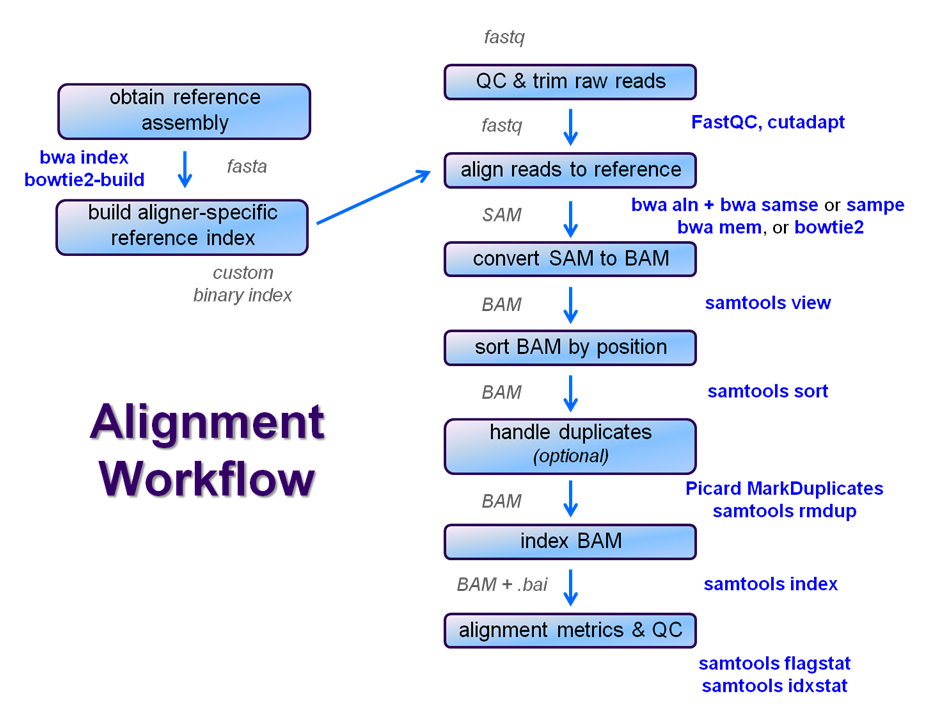 Image Added
Image Added
After Once raw sequence files are generated (in FASTQ format) and , quality-checked, and pre-processed in some way, the next step in most many NGS pipelines is mapping to a reference genome.
For individual sequences of interest, it is common to use a tool like BLAST to identify genes or species of origin. However , a typical example normal NGS dataset may will have tens to hundreds of millions of readssequences, which BLAST and similar tools are not designed to handle. Thus , a large set of computational tools have been developed to quickly , and with sufficient (but not absolute) accuracy align each read to its best location , (if any, ) in a reference.
Even though many mapping tools exist, a few individual programs have a dominant "market share" of the NGS world. These programs vary widely in their design, inputs, outputs, and applications. In this section, we will primarily focus on two of the most versatile mappersgeneral-purpose ones: BWA and Bowtie2, (the latter being part of the Tuxedo suite (e.g. which includes the transcriptome-aware RNA-seq aligner Tophat2 as well as other downstream quantifiaction tools).
Sample Datasets
Stage the alignment data
First connect to stampede2.tacc.utexas.edu and start an idev session. This should be second nature by now  You have already worked with a paired-end yeast ChIP-seq dataset, which we will continue to use here. The paired end data should already be located at:
You have already worked with a paired-end yeast ChIP-seq dataset, which we will continue to use here. The paired end data should already be located at:
| Code Block |
|---|
| language | bash |
|---|
| title | "Permanent" location for original data |
|---|
|
$WORK/archive/original/2014_05.core_ngs |
We will also use two additional RNA-seq datasets, which are located at:
| Code Block |
|---|
$CLASSDIR/human_stuff |
|
idev -p normal -m 180 -A UT-2015-05-18 -N 1 -n 68 |
Then stage the sample datasets and references we will use.
| Code Block |
|---|
| language | bash |
|---|
| title | Get the alignment exercises files |
|---|
|
mkdir -p $SCRATCH/core_ngs/references/fasta
mkdir -p $SCRATCH/core_ngs/alignment/fastq
cp $CORENGS/references/*.fa $SCRATCH/core_ngs/references/fasta/
cp $CORENGS/alignment/*fastq.gz $SCRATCH/core_ngs/alignment/fastq/
cd $SCRATCH/core_ngs/alignment/fastq |
These are descriptions of the FASTQ files we copied:
| File Name | Description | Sample |
|---|
| Sample_Yeast_L005_R1.cat.fastq.gz | Paired-end Illumina, First of pair |
| File Name | Description | Sample |
|---|
Sample_Yeast_L005_R1.cat.fastq.gz | Paired-end Illumina, First of pair| , FASTQ | Yeast ChIP-seq |
| Sample_Yeast_L005_R2.cat.fastq.gz | Paired-end Illumina, Second of pair, FASTQ | Yeast ChIP-seq |
| human_rnaseq.fastq.gz | Paired-end Illumina, First of pair only, FASTQ | Human RNA-seq |
| human_mirnaseq.fastq.gz | Single-end Illumina, FASTQ | Human microRNA-seq |
First copy the two human datasets to your $SCRATCH/core_ngs/fastq_prep directory.
| Code Block |
|---|
| language | bash |
|---|
| title | Stage human FASTQ data |
|---|
|
cd $SCRATCH/core_ngs/fastq_prep
cp $CLASSDIR/human_stuff/*rnaseq.fastq.gz . |
Create a $SCRATCH/core_ngs/align directory and make a link to the fastq_prep directory.
| Code Block |
|---|
| language | bash |
|---|
| title | Prepare align directory |
|---|
|
mkdir -p $SCRATCH/core_ngs/align
cd $SCRATCH/core_ngs/align
ln -s -f ../fastq_prep fq
ls -l
ls fq |
Reference Genomes
Before we get to alignment, we need a genome to align to. We will use three different references here:
- the human genome (hg19)
- the yeast genome (sacCer3)
- and mirbase (v20), human subset
Mirbase is a collection of all known microRNAs in all species. We will use the human subset of that database as our alignment reference. This has the advantage of being significantly smaller than the human genome, while containing all the sequences we are actually interested in.
| Expand |
|---|
| title | If it's simpler and faster, would one ever want to align a miRNA dataset to hg19 rather than mirbase? If so, why? |
|---|
|
- Due to natural variation, sequencing errors, and processing issues, variation between reference sequence and sample sequence is always possible. Alignment to the human genome allows a putative "microRNA" read the opportunity to find a better alignment in a region of the genome that is not an annotated microRNA. If this occurs, we might think that a read represents a microRNA (since it aligned in the mirbase alignment), when it is actually more likely to have come from a non-miRNA area of the genome.
- If we suspect our library contained other RNA species, we may want to quantify the level of "contamination". Aligning to the human genome will allow rRNA, tRNA, snoRNA, etc to align. We can then use programs such as bedtools, coupled with appropriate genome annotation files, to quantify these "off-target" hits.
|
These are the three reference genomes we will be using today, with some information about them (and here is information about many more genomes):
Searching genomes is hard work and takes a long time if done on an un-indexed, linear genomic sequence. So aligners require that references first be indexed for quick access The aligners we are using each require a different index, but use the same method (the Burrows-Wheeler Transform) to get the job done. This requires taking a FASTA file as input, with each chromosome (or contig) as a separate entry, and producing some aligner-specific set of files as output. Those index files are then used by the aligner when performing the sequence alignment.
hg19 is way too big for us to index here, so we're not going to do it. Instead, all hg19 index files are located at:
| Code Block |
|---|
| language | bash |
|---|
| title | BWA hg19 index location |
|---|
|
/scratch/01063/abattenh/ref_genome/bwa/bwtsw/hg19 |
We will grab the FASTA files for the other two references and build each index right before we use. These two references are located at:
| Code Block |
|---|
| language | bash |
|---|
| title | Yeast and mirbase FASTA locations |
|---|
|
/corral-repl/utexas/BioITeam/core_ngs_tools/references/sacCer3.fa
/corral-repl/utexas/BioITeam/core_ngs_tools/references/hairpin_cDNA_hsa.fa |
First stage the yeast and mirbase reference FASTA files in your work archive area in a directory called references.
| Code Block |
|---|
| language | bash |
|---|
| title | Stage FASTA references |
|---|
|
mkdir -p $WORK/archive/references/fasta
cp $CLASSDIR/references/*.fa $WORK/archive/references/fasta/ |
With that, we're ready to get started on the first exercise.
Exercise #1: BWA – Yeast ChIP-seq
Overview ChIP-seq alignment workflow with BWA
We will perform a global alignment of the paired-end Yeast ChIP-seq sequences using bwa. This workflow generally has the following steps:
- Trim the FASTQ sequences down to 50 with fastx_clipper
- this removes most of any 5' adapter contamination without the fuss of specific adapter trimming w/cutadapt
- Prepare the sacCer3 reference index for bwa (one time) using bwa index
- Perform a global bwa alignment on the R1 reads (bwa aln) producing a BWA-specific binary .sai intermediate file
- Perform a global bwa alignment on the R2 reads (bwa aln) producing a BWA-specific binary .sai intermediate file
- Perform pairing of the separately aligned reads and report the alignments in SAM format using bwa sampe
- Convert the SAM file to a BAM file (samtools view)
- Sort the BAM file by genomic location (samtools sort)
- Index the BAM file (samtools index)
- Gather simple alignment statistics (samtools flagstat and samtools idxstat)
We're going to skip the trimming step for now and see how it goes. We'll perform steps 2 - 5 now and leave samtools for the next course section, since those steps (6 - 10) are common to nearly all post-alignment workflows.
Introducing BWA
Like other tools you've worked with so far, you first need to load bwa using the module system. Go ahead and do that now, and then enter bwa with no arguments to view the top-level help page (many NGS tools will provide some help when called with no arguments).
| Code Block |
|---|
module load bwa
bwa |
...
...
| cholera_rnaseq.fastq.gz | Single-end Illumina, FASTQ | V. cholerae RNA-seq |
Reference Genomes
Before we get to alignment, we need a reference to align to. This is usually an organism's genome, but can also be any set of names sequences, such as a transcriptome or other set of genes.
Here are the four reference genomes we will be using today, with some information about them. These are not necessarily the most recent versions of these references (e.g. the newest human reference genome is hg38 and the most recent miRBase annotation is v21. (See here for information about many more genomes.)
Searching genomes is computationally hard work and takes a long time if done on linear genomic sequence. So aligners require that references first be indexed to accelerate lookup. The aligners we are using each require a different index, but use the same method (the Burrows-Wheeler Transform) to get the job done.
Building a reference index involves taking a FASTA file as input, with each contig (contiguous string of bases, e.g. a chromosome) as a separate FASTA entry, and producing an aligner-specific set of files as output. Those output index files are then used to perform the sequence alignment, and alignments are reported using coordinates referencing names and offset positions based on the original FASTA file contig entries.
We can quickly index the references for the yeast genome, the human miRNAs, and the V. cholerae genome, because they are all small, so we'll build each index from the appropriate FASTA files right before we use them.
hg19 is way too big for us to index here so we will use an existing set of BWA hg19 index files located at:
| Code Block |
|---|
| language | bash |
|---|
| title | BWA hg19 index location |
|---|
|
/work2/projects/BioITeam/ref_genome/bwa/bwtsw/hg19 |
| Tip |
|---|
The BioITeam maintains a set of reference indexes for many common organisms and aligners. They can be found in aligner-specific sub-directories of the /work2/projects/BioITeam/ref_genome area. E.g.: | Code Block |
|---|
| /work2/projects/BioITeam/ref_genome/
bowtie2/
bwa/
hisat2/
kallisto/
star/
tophat/ |
|
Exploring FASTA with grep
It is often useful to know what chromosomes/contigs are in a FASTA file before you start an alignment so that you're familiar with the contig naming convention – and to verify that it's the one you expect. For example, chromosome 1 is specified differently in different references and organisms: chr1 (USCS human), chrI (UCSC yeast), or just 1 (Ensembl human GRCh37).
A FASTA file consists of a number of contig name entries, each one starting with a right carat ( > ) character, followed by many lines of base characters. E.g.:
| Code Block |
|---|
>chrI
CCACACCACACCCACACACCCACACACCACACCACACACCACACCACACC
CACACACACACATCCTAACACTACCCTAACACAGCCCTAATCTAACCCTG
GCCAACCTGTCTCTCAACTTACCCTCCATTACCCTGCCTCCACTCGTTAC
CCTGTCCCATTCAACCATACCACTCCGAACCACCATCCATCCCTCTACTT |
How do we dig out just the lines that have the contig names and ignore all the sequences? Well, the contig name lines all follow the pattern above, and since the > character is not a valid base, it will never appear on a sequence line.
We've discovered a pattern (also known as a regular expression) to use in searching, and the command line tool that does regular expression matching is grep (general regular expression parser). Read more about grep here: Advanced commands: grep.
Regular expressions are so powerful that nearly every modern computer language includes a "regex" module of some sort. There are many online tutorials for regular expressions, and several slightly different "flavors" of them. But the most common is the Perl style (http://perldoc.perl.org/perlretut.html), which was one of the fist and still the most powerful (there's a reason Perl was used extensively when assembling the human genome). We're only going to use the most simple of regular expressions here, but learning more about them will pay handsome dividends for you in the future.
Here's how to execute grep to list contig names in a FASTA file.
| Code Block |
|---|
| language | bash |
|---|
| title | grep to match contig names in a FASTA file |
|---|
|
cd $SCRATCH/core_ngs/references/fasta
grep -P '^>' sacCer3.fa | more |
Notes:
- The -P option tells grep to Perl-style regular expression patterns.
- This makes including special characters like Tab ( \t ), Carriage Return ( \r ) or Linefeed ( \n ) much easier that the default POSIX paterns.
- While it is not required here, it generally doesn't hurt to include this option.
'^>' is the regular expression describing the pattern we're looking for (described below)
- sacCer3.fa is the file to search.
- lines with text that match our pattern will be written to standard output
- non matching lines will be omitted
- We pipe to more just in case there are a lot of contig names.
Now down to the nuts and bolts of the pattern: '^>'
First, the single quotes around the pattern – this tells the bash shell to pass the exact string contents to grep.
As part of its friendly command line parsing and evaluation, the shell will often look for special characters on the command line that mean something to it (for example, the $ in front of an environment variable name, like in $SCRATCH). Well, regular expressions treat the $ specially too – but in a completely different way! Those single quotes tell the shell "don't look inside here for special characters – treat this as a literal string and pass it to the program". The shell will obey, will strip the single quotes off the string, and will pass the actual pattern, ^>, to the grep program. (Note that the shell does look inside double quotes ( " ) for certain special signals, such as looking for environment variable names to evaluate. Read more about Quoting in the shell.)
So what does ^> mean to grep? We know that contig name lines always start with a > character, so > is a literal for grep to use in its pattern match.
We might be able to get away with just using this literal alone as our regex, specifying '>' as the command line argument. But for grep, the more specific the pattern, the better. So we constrain where the > can appear on the line. The special carat ( ^ ) metacharacter represents "beginning of line". So ^> means "beginning of a line followed by a > character".
Exercise: How many contigs are there in the sacCer3 reference?
| Expand |
|---|
|
| Code Block |
|---|
| language | bash |
|---|
| title | Get the alignment exercises files |
|---|
| mkdir -p $SCRATCH/core_ngs/references/fasta
cp $CORENGS/references/*.fa $SCRATCH/core_ngs/references/fasta/ |
|
| Expand |
|---|
|
| Code Block |
|---|
| cd $SCRATCH/core_ngs/references/fasta
grep -P '^>' sacCer3.fa | wc -l |
Or use grep's -c option that says "just count the line matches" | Code Block |
|---|
| grep -P -c '^>' sacCer3.fa |
|
| Expand |
|---|
|
There are 17 contigs. |
Aligner overview
There are many aligners available, but we will concentrate on two of the most popular general-purpose ones: bwa and bowtie2. The table below outlines the available protocols for them.
| alignment type | aligner options | pro's | con's |
|---|
| global with bwa | SE: PE: - bwa aln <R1>
- bwa aln <R2>
- bwa sampe
| - simple to use (take default options)
- good for basic global alignment
| |
| global with bowtie2 | bowtie2 --global | - extremely configurable
- can be used for RNAseq alignment (after adapter trimming) because of its many options
| |
| local with bwa | bwa mem | - simple to use (take default options)
- very fast
- no adapter trimming needed
- good for simple RNAseq analysis
- the secondary alignments it reports provide splice junction information
| - always produces alignments with secondary reads
- must be filtered if not desired
|
| local with bowtie2 | bowtie2 --local | - extremely configurable
- no adapter trimming needed
- good for small RNA alignment because of its many options
| |
Exercise #1: BWA global alignment – Yeast ChIP-seq
Overview ChIP-seq alignment workflow with BWA
We will perform a global alignment of the paired-end Yeast ChIP-seq sequences using bwa. This workflow has the following steps:
- Trim the FASTQ sequences down to 50 with fastx_clipper
- this removes most of any 5' adapter contamination without the fuss of specific adapter trimming w/cutadapt
- Prepare the sacCer3 reference index for bwa using bwa index
- this is done once, and re-used for later alignments
- Perform a global bwa alignment on the R1 reads (bwa aln) producing a BWA-specific binary .sai intermediate file
- Perform a global bwa alignment on the R2 reads (bwa aln) producing a BWA-specific binary .sai intermediate file
- Perform pairing of the separately aligned reads and report the alignments in SAM format using bwa sampe
- Convert the SAM file to a BAM file (samtools view)
- Sort the BAM file by genomic location (samtools sort)
- Index the BAM file (samtools index)
- Gather simple alignment statistics (samtools flagstat and samtools idxstat)
We're going to skip the trimming step for now and see how it goes. We'll perform steps 2 - 5 now and leave samtools for a later exercise since steps 6 - 10 are common to nearly all post-alignment workflows.
Introducing BWA
Like other tools you've worked with so far, you first need to load bwa. Do that now, and then enter bwa with no arguments to view the top-level help page (many NGS tools will provide some help when called with no arguments). Note that bwa is available both from the standard TACC module system and as BioContainers. module.
| Expand |
|---|
| title | Make sure you're in a idev session |
|---|
|
| Code Block |
|---|
| language | bash |
|---|
| title | Start an idev session |
|---|
| idev -p normal -m 120 -A UT-2015-05-18 -N 1 -n 68 |
|
| Code Block |
|---|
|
module load biocontainers # takes a while
module load bwa
bwa |
| Code Block |
|---|
|
Program: bwa (alignment via Burrows-Wheeler transformation)
Version: 0.7.17-r1188
Contact: Heng Li <lh3@sanger.ac.uk>
Usage: bwa <command> [options]
Command: index index sequences in the FASTA format
mem BWA-MEM algorithm
fastmap identify super-maximal exact matches
pemerge merge overlapping paired ends (EXPERIMENTAL)
aln gapped/ungapped alignment
samse generate alignment (single ended)
sampe generate alignment (paired ended)
bwasw BWA-SW for long queries
shm manage indices in shared memory
fa2pac convert FASTA to PAC format
pac2bwt generate BWT from PAC
pac2bwtgen alternative algorithm for generating BWT
bwtupdate update .bwt to the new format
bwt2sa generate SA from BWT and Occ
Note: To use BWA, you need to first index the genome with `bwa index'.
There are three alignment algorithms in BWA: `mem', `bwasw', and
`aln/samse/sampe'. If you are not sure which to use, try `bwa mem'
first. Please `man ./bwa.1' for the manual. |
As you can see, bwa include many sub-commands that perform the tasks we are interested in.
Building the BWA sacCer3 index
We will index the genome with the bwa index command. Type bwa index with no arguments to see usage for this sub-command.
| Code Block |
|---|
|
Usage: bwa index [options] <in.fasta>
Options: -a STR BWT construction algorithm: bwtsw, is or rb2 [auto]
-p STR prefix of the index [same as fasta name]
-b INT block size for the bwtsw algorithm (effective with -a bwtsw) [10000000]
-6 index files named as <in.fasta>.64.* instead of <in.fasta>.*
Warning: `-a bwtsw' does not work for short genomes, while `-a is' and
`-a div' do not work not for long genomes. |
Based on the usage description, we only need to specify two things:
- The name of the FASTA file
- Whether to use the bwtsw or is algorithm for indexing
Since sacCer3 is relative large (~12 Mbp) we will specify bwtsw as the indexing option (as indicated by the "Warning" message), and the name of the FASTA file is sacCer3.fa.
The output of this command is a group of files that are all required together as the index. So, within our references directory, we will create another directory called references/bwa/sacCer3 and build the index there. To remind ourselves which FASTA was used to build the index, we create a symbolic link to our references/fasta/sacCer3.fa file (note the use of the ../.. relative path syntax).
| Expand |
|---|
|
| Code Block |
|---|
| language | bash |
|---|
| title | Get the alignment exercises files |
|---|
| mkdir -p $SCRATCH/core_ngs/alignment/fastq
mkdir -p $SCRATCH/core_ngs/references/fasta
cp $CORENGS/alignment/*fastq.gz $SCRATCH/core_ngs/alignment/fastq/
cp $CORENGS/references/*.fa $SCRATCH/core_ngs/references/fasta/ |
|
| Code Block |
|---|
| language | bash |
|---|
| title | Prepare BWA reference directory for sacCer3 |
|---|
|
mkdir -p $SCRATCH/core_ngs/references/bwa/sacCer3
cd $SCRATCH/core_ngs/references/bwa/sacCer3
ln -s ../../fasta/sacCer3.fa
ls -l |
Now execute the bwa index command.
| Code Block |
|---|
| language | bash |
|---|
| title | Build BWA index for sacCer3 |
|---|
|
bwa index -a bwtsw sacCer3.fa |
Since the yeast genome is not large when compared to human, this should not take long to execute (otherwise we would do it as a batch job). When it is complete you should see a set of index files like this:
| Code Block |
|---|
| title | BWA index files for sacCer3 |
|---|
|
sacCer3.fa
sacCer3.fa.amb
sacCer3.fa.ann
sacCer3.fa.bwt
sacCer3.fa.pac
sacCer3.fa.sa |
Performing the bwa alignment
Now, we're ready to execute the actual alignment, with the goal of initially producing a SAM file from the input FASTQ files and reference. First prepare a directory for this exercise and link the sacCer3 reference directories there (this will make our commands more readable).
| Expand |
|---|
|
| Code Block |
|---|
| # Copy the pre-built references
mkdir -p $SCRATCH/core_ngs/references
cp $CORENGS/references/*.fa $SCRATCH/core_ngs/references/fasta/
# Get the FASTQ to align
mkdir -p $SCRATCH/core_ngs/alignment/fastq
cp $CORENGS/alignment/*fastq.gz $SCRATCH/core_ngs/alignment/fastq/
|
|
| Code Block |
|---|
| language | bash |
|---|
| title | Prepare to align yeast data |
|---|
|
mkdir -p $SCRATCH/core_ngs/alignment/yeast_bwa
cd $SCRATCH/core_ngs/alignment/yeast_bwa
ln -s -f ../fastq
ln -s -f ../../references/bwa/sacCer3 |
As our workflow indicated, we first use bwa aln on the R1 and R2 FASTQs, producing a BWA-specific .sai intermediate binary files.
What does bwa aln needs in the way of arguments?
There are lots of options, but here is a summary of the most important ones.
| Option | Effect |
|---|
| -l | Specifies the length of the seed (default = 32) |
| -k | Specifies the number of mismatches allowable in the seed of each alignment (default = 2) |
| -n | Specifies the number of mismatches (or fraction of bases in a given alignment that can be mismatches) in the entire alignment (including the seed) (default = 0.04) |
| -t | Specifies the number of threads |
Other options control the details of how much a mismatch or gap is penalized, limits on the number of acceptable hits per read, and so on. Much more information can be found on the BWA manual page.
For a basic alignment like this, we can just go with the default alignment parameters.
Note that bwa writes its (binary) output to standard output by default, so we need to redirect that to a .sai file.
For simplicity, we will just execute these commands directly, one at a time. Each command should only take few minutes and you will see bwa's progress messages in your terminal.
| Code Block |
|---|
| language | bash |
|---|
| title | bwa aln commands for yeast R1 and R2 |
|---|
|
# If not already loaded:
module load biocontainers
module load bwa
cd $SCRATCH/core_ngs/alignment/yeast_bwa
bwa aln sacCer3/sacCer3.fa fastq/Sample_Yeast_L005_R1.cat.fastq.gz > yeast_R1.sai
bwa aln sacCer3/sacCer3.fa fastq/Sample_Yeast_L005_R2.cat.fastq.gz > yeast_R2.sai |
When all is done you should have two .sai files: yeast_R1.sai and yeast_R2.sai.
| Tip |
|---|
| title | Make sure your output files are not empty |
|---|
|
Double check that output was written by doing ls -lh and making sure the file sizes listed are not 0. |
Exercise: How long did it take to align the R2 file?
| Expand |
|---|
|
The last few lines of bwa's execution output should look something like this: | Code Block |
|---|
| [bwa_aln] 17bp reads: max_diff = 2
[bwa_aln] 38bp reads: max_diff = 3
[bwa_aln] 64bp reads: max_diff = 4
[bwa_aln] 93bp reads: max_diff = 5
[bwa_aln] 124bp reads: max_diff = 6
[bwa_aln] 157bp reads: max_diff = 7
[bwa_aln] 190bp reads: max_diff = 8
[bwa_aln] 225bp reads: max_diff = 9
[bwa_aln_core] calculate SA coordinate... 50.76 sec
[bwa_aln_core] write to the disk... 0.07 sec
[bwa_aln_core] 262144 sequences have been processed.
[bwa_aln_core] calculate SA coordinate... 50.35 sec
[bwa_aln_core] write to the disk... 0.07 sec
[bwa_aln_core] 524288 sequences have been processed.
[bwa_aln_core] calculate SA coordinate... 13.64 sec
[bwa_aln_core] write to the disk... 0.01 sec
[bwa_aln_core] 592180 sequences have been processed.
[main] Version: 0.7.17-r1188
[main] CMD: /usr/local/bin/bwa aln sacCer3/sacCer3.fa fastq/Sample_Yeast_L005_R1.cat.fastq.gz
[main] Real time: 122.936 sec; CPU: 123.597 sec |
So the R2 alignment took ~123 seconds (~2 minutes). |
Since you have your own private compute node, you can use all its resources. It has 68 cores, so re-run the R2 alignment asking for 60 execution threads.
| Code Block |
|---|
bwa aln -t 60 sacCer3/sacCer3.fa fastq/Sample_Yeast_L005_R2.cat.fastq.gz > yeast_R2.sai |
Exercise: How much of a speedup did you seen when aligning the R2 file with 20 threads?
| Expand |
|---|
|
The last few lines of bwa's execution output should look something like this: | Code Block |
|---|
| [bwa_aln] 17bp reads: max_diff = 2
[bwa_aln] 38bp reads: max_diff = 3
[bwa_aln] 64bp reads: max_diff = 4
[bwa_aln] 93bp reads: max_diff = 5
[bwa_aln] 124bp reads: max_diff = 6
[bwa_aln] 157bp reads: max_diff = 7
[bwa_aln] 190bp reads: max_diff = 8
[bwa_aln] 225bp reads: max_diff = 9
[bwa_aln_core] calculate SA coordinate... 266.70 sec
[bwa_aln_core] write to the disk... 0.04 sec
[bwa_aln_core] 262144 sequences have been processed.
[bwa_aln_core] calculate SA coordinate... 268.94 sec
[bwa_aln_core] write to the disk... 0.03 sec
[bwa_aln_core] 524288 sequences have been processed.
[bwa_aln_core] calculate SA coordinate... 72.26 sec
[bwa_aln_core] write to the disk... 0.01 sec
[bwa_aln_core] 592180 sequences have been processed.
[main] Version: 0.7.17-r1188
[main] CMD: /usr/local/bin/bwa aln -t 60 sacCer3/sacCer3.fa fastq/Sample_Yeast_L005_R2.cat.fastq.gz
[main] Real time: 19.872 sec; CPU: 617.095 sec |
So the R2 alignment took only ~20 seconds (real time), or 6+ times as fast as with only one processing thread. Note, though, that the CPU time with 60 threads was greater (617 sec) than with only 1 thread (124 sec). That's because of the thread management overhead when using multiple threads. |
Next we use the bwa sampe command to pair the reads and output SAM format data. Just type that command in with no arguments to see its usage.
For this command you provide the same reference index prefix as for bwa aln, along with the two .sai files and the two original FASTQ files. Also, bwa writes its output to standard output, so redirect that to a .sam file.
Here is the command line statement you need. Just execute it on the command line.
| Code Block |
|---|
| language | bash |
|---|
| title | Pairing of BWA R1 and R2 aligned reads |
|---|
|
bwa sampe sacCer3/sacCer3.fa yeast_R1.sai yeast_R2.sai \
fastq/Sample_Yeast_L005_R1.cat.fastq.gz \
fastq/Sample_Yeast_L005_R2.cat.fastq.gz > yeast_pairedend.sam |
You should now have a SAM file (yeast_pairedend.sam) that contains the alignments. It's just a text file, so take a look with head, more, less, tail, or whatever you feel like. Later you'll learn additional ways to analyze the data with samtools once you create a BAM file.
Exercise: What kind of information is in the first lines of the SAM file?
| Expand |
|---|
|
The SAM file has a number of header lines, which all start with an at sign ( @ ). The @SQ lines describe each contig (chromosome) and its length. There is also a @PG line that describes the way the bwa sampe was performed. |
Exercise: How many alignment records (not header records) are in the SAM file?
| Expand |
|---|
|
This looks for the pattern '^HWI' which is the start of every read name (which starts every alignment record).
Remember -c says just count the records, don't display them. | Code Block |
|---|
| grep -P -c '^HWI' yeast_pairedend.sam |
Or use the -v (invert) option to tell grep to print all lines that don't match a particular pattern; here, all header lines, which start with @. | Code Block |
|---|
| grep -P -v -c '^@' yeast_pairedend.sam |
|
| Expand |
|---|
|
| There are 1,184,360 alignment records. |
Exercise: How many sequences were in the R1 and R2 FASTQ files combined?
| Expand |
|---|
|
zcat fastq/Sample_Yeast_L005_R[12].cat.fastq.gz | wc -l | awk '{print $1/4}'
|
| Expand |
|---|
|
| There were a total of 1,184,360 original sequences (R1s + R2s) |
Exercises:
- Do both R1 and R2 reads have separate alignment records?
- Does the SAM file contain both mapped and un-mapped reads?
- What is the order of the alignment records in this SAM file?
| Expand |
|---|
|
Both R1 and R2 reads must have separate alignment records, because there were 1,184,360 R1+R2 reads and the same number of alignment records. The SAM file must contain both mapped and un-mapped reads, because there were 1,184,360 R1+R2 reads and the same number of alignment records. Alignment records occur in the same read-name order as they did in the FASTQ, except that they come in pairs. The R1 read comes 1st, then the corresponding R2. This is called read name ordering. |
Using cut to isolate fields
Recall the format of a SAM alignment record:
 Image Added
Image Added
Suppose you wanted to look only at field 3 (contig name) values in the SAM file. You can do this with the handy cut command. Below is a simple example where you're asking cut to display the 3rd column value for the last 10 alignment records.
| Code Block |
|---|
| language | bash |
|---|
| title | Cut syntax for a single field |
|---|
|
tail yeast_pairedend.sam | cut -f 3 |
By default cut assumes the field delimiter is Tab, which is the delimiter used in the majority of NGS file formats. You can specify a different delimiter with the -d option.
You can also specify a range of fields, and mix adjacent and non-adjacent fields. This displays fields 2 through 6, field 9:
| Code Block |
|---|
| language | bash |
|---|
| title | Cut syntax for multiple fields |
|---|
|
tail -20 yeast_pairedend.sam | cut -f 2-6,9 |
You may have noticed that some alignment records contain contig names (e.g. chrV) in field 3 while others contain an asterisk ( * ). The * means the record didn't map. We're going to use this heuristic along with cut to see about how many records represent aligned sequences. (Note this is not the strictly correct method of finding unmapped reads because not all unmapped reads have an asterisk in field 3. Later you'll see how to properly distinguish between mapped and unmapped reads using samtools.)
First we need to make sure that we don't look at fields in the SAM header lines. We're going to end up with a series of pipe operations, and the best way to make sure you're on track is to enter them one at a time piping to head:
| Code Block |
|---|
| language | bash |
|---|
| title | Grep pattern that doesn't match header |
|---|
|
# the ^@ pattern matches lines starting with @ (only header lines),
# and -v says output lines that don't match
grep -v -P '^@' yeast_pairedend.sam | head |
Ok, it looks like we're seeing only alignment records. Now let's pull out only field 3 using cut:
| Code Block |
|---|
| language | bash |
|---|
| title | Get contig name info with cut |
|---|
|
grep -v -P '^@' yeast_pairedend.sam | cut -f 3 | head |
Cool, we're only seeing the contig name info now. Next we use grep again, piping it our contig info and using the -v (invert) switch to say print lines that don't match the pattern:
| Code Block |
|---|
| language | bash |
|---|
| title | Filter contig name of * (unaligned) |
|---|
|
grep -v -P '^@' yeast_pairedend.sam | cut -f 3 | grep -v '*' | head |
Perfect! We're only seeing real contig names that (usually) represent aligned reads. Let's count them by piping to wc -l (and omitting omit head of course – we want to count everything).
| Code Block |
|---|
| language | bash |
|---|
| title | Count aligned SAM records |
|---|
|
grep -v -P '^@' yeast_pairedend.sam | cut -f 3 | grep -v '*' | wc -l |
Exercise: About how many records represent aligned sequences? What alignment rate does this represent?
| Expand |
|---|
|
The expression above returns 612,968. There were 1,184,360 records total, so the percentage is: | Code Block |
|---|
| language | bash |
|---|
| title | Calculate alignment rate |
|---|
| awk 'BEGIN{print 612968/1184360}' |
or about 51%. Not great. Note we perform this calculation in awk's BEGIN block, which is always executed, instead of the body block, which is only executed for lines of input. And here we call awk without piping it any input. See Linux fundamentals: cut,sort,uniq,grep,awk |
Exercise: What might we try in order to improve the alignment rate?
| Expand |
|---|
|
| Recall that these are 100 bp reads and we did not remove adapter contamination. There will be a distribution of fragment sizes – some will be short – and those short fragments may not align without adapter removal (e.g. with fastx_trimmer). |
Exercise #2: Basic SAMtools Utilities
The SAMtools program is a commonly used set of tools that allow a user to manipulate SAM/BAM files in many different ways, ranging from simple tasks (like SAM/BAM format conversion) to more complex functions (like sorting, indexing and statistics gathering). It is available in the TACC module system (as well as in BioContainers). Load that module and see what samtools has to offer:
| Expand |
|---|
| title | Make sure you're in a idev session |
|---|
|
| Code Block |
|---|
| language | bash |
|---|
| title | Start an idev session |
|---|
| idev -p normal -m 120 -A UT-2015-05-18 -N 1 -n 68 |
|
| Code Block |
|---|
|
# If not already loaded
module load biocontainers # takes a while
module load samtools
samtools |
| Code Block |
|---|
| title | SAMtools suite usage |
|---|
|
Program: samtools (Tools for alignments in the SAM format)
Version: 1.10 (using htslib 1.10)
Usage: samtools <command> [options]
Commands:
-- Indexing
dict create a sequence dictionary file
faidx index/extract FASTA
fqidx index/extract FASTQ
index index alignment
-- Editing
calmd recalculate MD/NM tags and '=' bases
fixmate fix mate information
reheader replace BAM header
targetcut cut fosmid regions (for fosmid pool only)
addreplacerg adds or replaces RG tags
markdup mark duplicates
-- File operations
collate shuffle and group alignments by name
cat concatenate BAMs
merge merge sorted alignments
mpileup multi-way pileup
sort sort alignment file
split splits a file by read group
quickcheck quickly check if SAM/BAM/CRAM file appears intact
fastq converts a BAM to a FASTQ
fasta converts a BAM to a FASTA
-- Statistics
bedcov read depth per BED region
coverage alignment depth and percent coverage
depth compute the depth
flagstat simple stats
idxstats BAM index stats
phase phase heterozygotes
stats generate stats (former bamcheck)
-- Viewing
flags explain BAM flags
tview text alignment viewer
view SAM<->BAM<->CRAM conversion
depad convert padded BAM to unpadded BAM |
In this exercise, we will explore five utilities provided by samtools: view, sort, index, flagstat, and idxstats. Each of these is executed in one line for a given SAM/BAM file. In the SAMtools/BEDtools sections tomorrow we will explore samtools in more in depth.
| Warning |
|---|
| title | Know your samtools version! |
|---|
|
There are two main "eras" of SAMtools development: - "original" samtools
- v 0.1.19 is the last stable version
- "modern" samtools
- v 1.0, 1.1, 1.2 – avoid these (very buggy!)
- v 1.3+ – finally stable!
Unfortunately, some functions with the same name in both version eras have different options and arguments! So be sure you know which version you're using. (The samtools version is usually reported at the top of its usage listing). TACC BioContainers also offers the original samtools version: samtools/ctr-0.1.19--3. |
samtools view
The samtools view utility provides a way of converting between SAM (text) and BAM (binary, compressed) format. It also provides many, many other functions which we will discuss lster. To get a preview, execute samtools view without any other arguments. You should see:
| Code Block |
|---|
|
Usage: samtools view [options] <in.bam>|<in.sam>|<in.cram> [region ...]
Options:
-b output BAM
-C output CRAM (requires -T)
-1 use fast BAM compression (implies -b)
-u uncompressed BAM output (implies -b)
-h include header in SAM output
-H print SAM header only (no alignments)
-c print only the count of matching records
-o FILE output file name [stdout]
-U FILE output reads not selected by filters to FILE [null]
-t FILE FILE listing reference names and lengths (see long help) [null]
-X include customized index file
-L FILE only include reads overlapping this BED FILE [null]
-r STR only include reads in read group STR [null]
-R FILE only include reads with read group listed in FILE [null]
-d STR:STR
only include reads with tag STR and associated value STR [null]
-D STR:FILE
only include reads with tag STR and associated values listed in
FILE [null]
-q INT only include reads with mapping quality >= INT [0]
-l STR only include reads in library STR [null]
-m INT only include reads with number of CIGAR operations consuming
query sequence >= INT [0]
-f INT only include reads with all of the FLAGs in INT present [0]
-F INT only include reads with none of the FLAGS in INT present [0]
-G INT only EXCLUDE reads with all of the FLAGs in INT present [0]
-s FLOAT subsample reads (given INT.FRAC option value, 0.FRAC is the
fraction of templates/read pairs to keep; INT part sets seed)
-M use the multi-region iterator (increases the speed, removes
duplicates and outputs the reads as they are ordered in the file)
-x STR read tag to strip (repeatable) [null]
-B collapse the backward CIGAR operation
-? print long help, including note about region specification
-S ignored (input format is auto-detected)
--no-PG do not add a PG line
--input-fmt-option OPT[=VAL]
Specify a single input file format option in the form
of OPTION or OPTION=VALUE
-O, --output-fmt FORMAT[,OPT[=VAL]]...
Specify output format (SAM, BAM, CRAM)
--output-fmt-option OPT[=VAL]
Specify a single output file format option in the form
of OPTION or OPTION=VALUE
-T, --reference FILE
Reference sequence FASTA FILE [null]
-@, --threads INT
Number of additional threads to use [0]
--write-index
Automatically index the output files [off]
--verbosity INT
Set level of verbosity |
That is a lot to process! For now, we just want to read in a SAM file and output a BAM file. The input format is auto-detected, so we don't need to specify it (although you do in v0.1.19). We just need to tell the tool to output the file in BAM format, and to include the header records.
| Expand |
|---|
|
| Code Block |
|---|
| language | bash |
|---|
| title | Get the alignment exercises files |
|---|
| mkdir -p $SCRATCH/core_ngs/alignment/yeast_bwa
cd $SCRATCH/core_ngs/alignment/yeast_bwa
cp $CORENGS/catchup/yeast_bwa/yeast_pairedend.sam .
|
|
| Code Block |
|---|
| language | bash |
|---|
| title | Convert SAM to binary BAM |
|---|
|
cd $SCRATCH/core_ngs/alignment/yeast_bwa
cat yeast_pairedend.sam | samtools view -b -o yeast_pairedend.bam |
- the -b option tells the tool to output BAM format
- the -o option specifies the name of the output BAM file that will be created
- we pipe the entire SAM file to samtools view so that the header records are included (required for SAM → BAM conversion)
- samtools view reads its input from standard input by default
How do you look at the BAM file contents now? That's simple. Just use samtools view without the -b option. Remember to pipe output to a pager!
| Code Block |
|---|
| language | bash |
|---|
| title | View BAM records |
|---|
|
samtools view yeast_pairedend.bam | more
|
Notice that this does not show us the header record we saw at the start of the SAM file.
Exercise: What samtools view option will include the header records in its output? Which option would show only the header records?
| Expand |
|---|
|
samtools view -h shows header records along with alignment records. samtools view -H shows header records only. |
samtools sort
Looking at some of the alignment record information (e.g. samtools view yeast_pairedend.bam | cut -f 1-4 | more), you will notice that read names appear in adjacent pairs (for the R1 and R2), in the same order they appeared in the original FASTQ file. Since that means the corresponding mappings are in no particular order, searching through the file very inefficient. samtools sort re-orders entries in the SAM file either by locus (contig name + coordinate position) or by read name.
If you execute samtools sort without any options, you see its help page:
| Code Block |
|---|
|
Usage: samtools sort [options...] [in.bam]
Options:
-l INT Set compression level, from 0 (uncompressed) to 9 (best)
-m INT Set maximum memory per thread; suffix K/M/G recognized [768M]
-n Sort by read name
-t TAG Sort by value of TAG. Uses position as secondary index (or read name if -n is set)
-o FILE Write final output to FILE rather than standard output
-T PREFIX Write temporary files to PREFIX.nnnn.bam
--no-PG do not add a PG line
--input-fmt-option OPT[=VAL]
Specify a single input file format option in the form
of OPTION or OPTION=VALUE
-O, --output-fmt FORMAT[,OPT[=VAL]]...
Specify output format (SAM, BAM, CRAM)
--output-fmt-option OPT[=VAL]
Specify a single output file format option in the form
of OPTION or OPTION=VALUE
--reference FILE
Reference sequence FASTA FILE [null]
-@, --threads INT
Number of additional threads to use [0]
--verbosity INT
Set level of verbosity |
In most cases you will be sorting a BAM file from name order to locus order. You can use either -o or redirection with > to control the output.
| Expand |
|---|
|
Copy aligned yeast BAM file | Code Block |
|---|
| mkdir -p $SCRATCH/core_ngs/alignment/yeast_bwa
cd $SCRATCH/core_ngs/alignment/yeast_bwa
cp $CORENGS/catchup/yeast_bwa/yeast_pairedend.bam . |
|
To sort the paired-end yeast BAM file by position, and get a BAM file named yeast_pairedend.sort.bam as output, execute the following command:
| Code Block |
|---|
| language | bash |
|---|
| title | Sort a BAM file |
|---|
|
cd $SCRATCH/core_ngs/alignment/yeast_bwa
samtools sort -O bam -T yeast_pairedend.tmp yeast_pairedend.bam > yeast_pairedend.sort.bam |
- The -O options says the Output format should be BAM
- The -T options gives a prefix for Temporary files produced during sorting
- sorting large BAMs will produce many temporary files during processing
- By default sort writes its output to standard output, so we use > to redirect to a file named yeast_pairedend.sort.bam
Exercise: Compare the file sizes of the yeast_pariedend .sam, .bam, and .sort.bam files and explain why they are different.
| Expand |
|---|
|
| Code Block |
|---|
| ls -lh yeast_pairedend* |
|
| Expand |
|---|
|
The yeast_pairedend.sam text file is the largest at ~348 MB. The name-ordered binary yeast_pairedend.bam text file only about 1/3 that size, ~110 MB. They contain exactly the same records, in the same order, but conversion from text to binary results in a much smaller file. The coordinate-ordered binary yeast_pairedend.sort.bam file is even slightly smaller, ~91 MB. This is because BAM files are actually customized gzip-format files. The customization allows blocks of data (e.g. all alignment records for a contig) to be represented in an even more compact form. You can read more about this in section 4 of the SAM format specification. |
samtools index
Many tools (like IGV, the Integrative Genomics Viewer) only need to use portions of a BAM file at a given point in time. For example, if you are viewing alignments that are within a particular gene, alignment records on other chromosomes do not need to be loaded. In order to speed up access, BAM files are indexed, producing BAI files which allow fast random access. This is especially important when you have many alignment records.
The utility samtools index creates an index that has the same name as the input BAM file, with suffix .bai appended. Here's the samtools index usage:
| Code Block |
|---|
| title | samtools index usage |
|---|
|
Usage: samtools index [-bc] [-m INT] <in.bam> [out.index]
Options:
-b Generate BAI-format index for BAM files [default]
-c Generate CSI-format index for BAM files
-m INT Set minimum interval size for CSI indices to 2^INT [14]
-@ INT Sets the number of threads [none] |
The syntax here is way, way easier. We want a BAI-format index which is the default. (CSI-format is used with extremely long contigs, which don't apply here - the most common use case is for polyploid plant genomes).
So all we have to provide is the sorted BAM:
| Code Block |
|---|
| language | bash |
|---|
| title | Index a sorted bam |
|---|
|
samtools index yeast_pairedend.sort.bam |
This will produce a file named yeast_pairedend.bam.bai.
Most of the time when an index is required, it will be automatically located as long as it is in the same directory as its BAM file and shares the same name up until the .bai extension.
Exercise: Compare the sizes of the sorted BAM file and its BAI index.
| Expand |
|---|
|
| Code Block |
|---|
| ls -lh yeast_pairedend.sort.bam* |
|
| Expand |
|---|
|
While the yeast_pairedend.sort.bam text file is ~91 MB, its index (yeast_pairedend.sort.bai) is only 20 KB. |
samtools flagstat
Since the BAM file contains records for both mapped and unmapped reads, just counting records doesn't provide information about the mapping rate of our alignment. The samtools flagstat tool provides a simple analysis of mapping rate based on the the SAM flag fields.
Here's how to run samtools flagstat and both see the output in the terminal and save it in a file – the samtools flagstat standard output is piped to tee, which both writes it to the specified file and sends it to its standard output:
| Code Block |
|---|
| language | bash |
|---|
| title | Run samtools flagstat using tee |
|---|
|
samtools flagstat yeast_pairedend.sort.bam | tee yeast_pariedend.flagstat.txt |
You should see something like this:
| Code Block |
|---|
| title | samtools flagstat output |
|---|
|
1184360 + 0 in total (QC-passed reads + QC-failed reads)
0 + 0 secondary
0 + 0 supplementary
0 + 0 duplicates
547664 + 0 mapped (46.24% : N/A)
1184360 + 0 paired in sequencing
592180 + 0 read1
592180 + 0 read2
473114 + 0 properly paired (39.95% : N/A)
482360 + 0 with itself and mate mapped
65304 + 0 singletons (5.51% : N/A)
534 + 0 with mate mapped to a different chr
227 + 0 with mate mapped to a different chr (mapQ>=5) |
Ignore the "+ 0" addition to each line - that is a carry-over convention for counting QA-failed reads that is no longer relevant.
The most important statistic is the mapping rate (here 46%) but this readout also allows you to verify that some common expectations (e.g. that about the same number of R1 and R2 reads aligned, and that most mapped reads are proper pairs) are met.
Exercise: What proportion of mapped reads were properly paired?
| Expand |
|---|
|
Divide the number of properly paired reads by the number of mapped reads: | Code Block |
|---|
| awk 'BEGIN{ print 473114 / 547664 }'
# or
echo $(( 473114 * 100 / 547664 ))
|
|
| Expand |
|---|
|
About 86% of mapped read were properly paired. This is actually a bit on the low side for ChIP-seq alignments which typically over 90%. |
samtools idxstats
More information about the alignment is provided by the samtools idxstats report, which shows how many reads aligned to each contig in your reference. Note that samtools idxstats must be run on a sorted, indexed BAM file.
| Code Block |
|---|
| language | bash |
|---|
| title | Use samtools idxstats to summarize mapped reads by contig |
|---|
|
samtools idxstats yeast_pairedend.sort.bam | tee yeast_pairedend.idxstats.txt |
| Code Block |
|---|
| language | bash |
|---|
| title | samtools idxstats output |
|---|
|
chrI 230218 8820 1640
chrII 813184 36616 4026
chrIII 316620 13973 1530
chrIV 1531933 72675 8039
chrV 576874 27466 2806
chrVI 270161 10866 1222
chrVII 1090940 50893 5786
chrVIII 562643 24672 3273
chrIX 439888 16246 1739
chrX 745751 31748 3611
chrXI 666816 28017 2776
chrXII 1078177 54783 10124
chrXIII 924431 40921 4556
chrXIV 784333 33070 3703
chrXV 1091291 48714 5150
chrXVI 948066 44916 5032
chrM 85779 3268 291
* 0 0 571392 |
The output has four tab-delimited columns:
- contig name
- contig length
- number of mapped reads
- number of unmapped reads
The reason that the "unmapped reads" field for named chromosomes is not zero is that the aligner may initially assign a potential mapping (contig name and start coordinate) to a read, but then mark it later as unampped if it does meet various quality thresholds.
| Tip |
|---|
If you're mapping to a non-genomic reference such as miRBase miRNAs or another set of genes (a transcriptome), samtools idxstats gives you a quick look at quantitative alignment results |
As you can see, bwa offers a number of sub-commands one can use with to do different things.
Building the BWA sacCer3 index
We're going to index the genome with the index command. To learn what this sub-command needs in the way of options and arguments, enter bwa index with no arguments.
| Code Block |
|---|
Usage: bwa index [-a bwtsw|is] [-c] <in.fasta>
Options: -a STR BWT construction algorithm: bwtsw or is [auto]
-p STR prefix of the index [same as fasta name]
-6 index files named as <in.fasta>.64.* instead of <in.fasta>.*
Warning: `-a bwtsw' does not work for short genomes, while `-a is' and
`-a div' do not work not for long genomes. Please choose `-a'
according to the length of the genome. |
Here, we only need to specify two things:
- the name of the FASTA file
- whether to use the bwtsw or is algorithm for indexing
Since sacCer3 is relative large (~12 Mbp) we will specify bwtsw as the indexing option, and the name of the FASTA file is sacCer3.fa.
Importantly, the output of this command is a group of files that are all required together as the index. So, within the references directory, we will create another directory called bwa/sacCer3, make a symbolic link to the yeast FASTA there, and run the index command in that directory.
| Code Block |
|---|
| language | bash |
|---|
| title | Prepare BWA reference directory for sacCer3 |
|---|
|
mkdir -p $WORK/archive/references/bwa/sacCer3
cd $WORK/archive/references/bwa/sacCer3
ln -s ../../fasta/sacCer3.fa
ls -la |
Now execute the bwa index command.
| Code Block |
|---|
| language | bash |
|---|
| title | Build BWA index for sacCer3 |
|---|
|
bwa index -a bwtsw sacCer3.fa |
Since the yeast genome is not large when compared to human, this should not take long to execute (otherwise we would do it as a batch job). When it is comple you should see a set of index files like this:
| Code Block |
|---|
| title | BWA index files for sacCer3 |
|---|
|
sacCer3.fa
sacCer3.fa.amb
sacCer3.fa.ann
sacCer3.fa.bwt
sacCer3.fa.pac
sacCer3.fa.sa |
Exploring the FASTA with grep
A common question is what contigs are in a given FASTA file. You'll usually want to know this before you start the alignment so that you're familiar with the contig naming convention – and to verify that it's the one you expect.
We saw that a FASTA consists of a number of contig entries, each one starting with a name line of the form below, followed by many lines of bases.
How do we dig out just the lines that have the contig names and ignore all the sequences? Well, the contig name lines all follow the pattern above, and since the > character is not a valid base, it will never appear on a sequence line.
We've discovered a pattern (also known as a regular expression) to use in searching, and the command line tool that does regular expression matching is grep.
Regular expressions are so powerful that nearly every modern computer language includes a "regex" module of some sort. There are many online tutorials for regular expressions (and a few different flavors of them). But the most common is the Perl style (http://perldoc.perl.org/perlretut.html). We're only going to use the most simple of regular expressions here, but learning more about them will pay handsome dividends for you in the future.
Here's how to execute grep to list contig names in a FASTA file.
| Code Block |
|---|
| language | bash |
|---|
| title | grep to match contig names in a FASTA file |
|---|
|
grep -P '^>' sacCer3.fa | more |
Notes:
- The -P option tells grep to use Perl-style regular expression patterns.
- This makes including special characters like Tab ( \t ), Carriage Return ( \r ) or Linefeed ( \n ) much easier that the default Posix paterns.
- While it is not really required here, it generally doesn't hurt to include this option.
'^>' is the regular expression describing the pattern we're looking for (described below)
- sacCer3.fa is the file to search. Lines with text that match our pattern will be written to standard output; non matching lines will be omitted.
- We pipe to more just in case there are a lot of contig names.
Now down to the nuts and bolts of our pattern, '^>'
First, the single quotes around the pattern – they are only a signal for the bash shell. As part of its friendly command line parsing and evaluation, the shell will often look for special characters on the command line that mean something to it (for example, the $ in front of an environment variable name, like in $SCRATCH). Well, regular expressions treat the $ specially too – but in a completely different way! Those single quotes tell the shell "don't look inside here for special characters – treat this as a literal string and pass it to the program". The shell will obey, will strip the single quotes off the string, and will pass the actual pattern, ^>, to the grep program. (Aside: We've see that the shell does look inside double quotes ( " ) for certain special signals, such as looking for environment variable names to evaluate.)
So what does ^> mean to grep? Well, from our contig name format description above we see that contig name lines always start with a > character, so > is a literal for grep to use in its pattern match.
We might be able to get away with just using this literal alone as our regex, specifying '>' as the command line argument. But for grep, the more specific the pattern, the better. So we constrain where the > can appear on the line. The special carat ( ^ ) character represents "beginning of line". So ^> means "beginning of a line followed by a > character, followed by anything. (Aside: the dollar sign ( $ ) character represents "end of line" in a regex. There are many other special characters, including period ( . ), question mark ( ? ), pipe ( | ), parentheses ( ( ) ), and brackets ( [ ] ), to name the most common.)
Exercise: How many contigs are there in the sacCer3 reference?
| Expand |
|---|
|
grep -P '^>' sacCer3.fa | wc -l
or use grep's -c option that says "just count the line matches" grep -P -c '^>' sacCer3.fa
|
| Expand |
|---|
|
There are 17 contigs. |
Performing the alignment
Now, we're ready to execute the actual alignment, with the goal of initially producing a SAM file from the input FASTQ files and reference. First go to the align directory, and link to the sacCer3 reference directory (this will make our commands more readable).
| Code Block |
|---|
| language | bash |
|---|
| title | Prepare to align yeast data |
|---|
|
cd $SCRATCH/core_ngs/align
ln -s $WORK/archive/references/bwa/sacCer3
ls sacCer3 |
As our workflow indicated, we first use bwa aln on the R1 and R2 FASTQs, producing a BWA-specific .sai intermediate binary files. Since these alignments are completely independent, we can execute them in parallel in a batch job.
What does bwa aln needs in the way of arguments?
There are lots of options, but here is a summary of the most important ones. BWA, is a lot more complex than the options let on. If you look at the BWA manual on the web for the aln sub-command, you'll see numerous options that can increase the alignment rate (as well as decrease it), and all sorts of other things.
| Option | Effect |
|---|
| -l | Controls the length of the seed (default = 32) |
| -k | Controls the number of mismatches allowable in the seed of each alignment (default = 2) |
| -n | Controls the number of mismatches (or fraction of bases in a given alignment that can be mismatches) in the entire alignment (including the seed) (default = 0.04) |
| -t | Controls the number of threads |
The rest of the options control the details of how much a mismatch or gap is penalized, limits on the number of acceptable hits per read, and so on. Much more information can be accessed at the BWA manual page.
For a simple alignment like this, we can just go with the default alignment parameters, with one exception. At TACC, we want to optimize our alignment speed by allocating more than one thread (-t) to the alignment. We want to run 2 tasks, and will use a minimum of one 16-core node. So we can assign 8 cores to each alignment by specifying -t 8.
Also note that bwa writes its (binary) output to standard output by default, so we need to redirect that to a .sai file. And of course we need to redirect standard error to a log file, one per file.
Create an aln.cmds file (using nano) with the following lines:
| Code Block |
|---|
| language | bash |
|---|
| title | bwa aln commands for yeast R1 and R2 |
|---|
|
bwa aln -t 8 sacCer3/sacCer3.fa fq/Sample_Yeast_L005_R1.cat.fastq.gz > yeast_R1.sai 2> aln.yeast_R1.log
bwa aln -t 8 sacCer3/sacCer3.fa fq/Sample_Yeast_L005_R2.cat.fastq.gz > yeast_R2.sai 2> aln.yeast_R2.log |
Create the batch submission script specifying a wayness of 8 (8 tasks per node) on the normal queue and a time of 1 hour, then submit the job and monitor the queue:
| Code Block |
|---|
| language | bash |
|---|
| title | Submit BWA alignment job |
|---|
|
launcher_creator.py -n aln -j aln.cmds -t 01:00:00 -q normal -w 8
sbatch aln.slurm
showq -u |
Since you have directed standard error to log files, you can use a neat trick to monitor the progress of the alignment: tail -f. The -f means "follow" the tail, so new lines at the end of the file are displayed as they are added to the file.
| Code Block |
|---|
| language | bash |
|---|
| title | Monitor alignment progress with tail -f |
|---|
|
# Use Ctrl-c to stop the output any time
tail -f aln.yeast_R1.log |
When it's done you should see two .sai files. Next we use the bwa sampe command to pair the reads and output SAM format data. For this command you provide the same reference prefix as for bwa aln, along with the two .sai files and the two original FASTQ files.
Again bwa writes its output to standard output, so redirect that to a .sam file. (Note that bwa sampe is "single threaded" – it does not have an option to use more than one processor for its work.) We'll just execute this at the command line – not in a batch job.
| Code Block |
|---|
| language | bash |
|---|
| title | BWA global alignment of R1 reads |
|---|
|
bwa sampe sacCer3/sacCer3.fa yeast_R1.sai yeast_R2.sai fq/Sample_Yeast_L005_R1.cat.fastq.gz fq/Sample_Yeast_L005_R2.cat.fastq.gz > yeast_pairedend.sam |
You did it! You should now have a SAM file that contains the alignments. It's just a text file, so take a look with head, more, less, tail, or whatever you feel like. In the next section, with samtools, you'll learn some additional ways to analyze the data once you create a BAM file.
Exercise: What kind of information is in the first lines of the SAM file?
| Expand |
|---|
|
| The SAM or BAM has a number of header lines, which all start with an at sign ( @ ). The @SQ lines describe each contig and its length. There is also a @PG line that describes the way the bwa sampe was performed. |
Exercise: How many alignment records (not header records) are in the SAM file?
| Expand |
|---|
|
This looks for the pattern '^HWI' which is the start of every read name (which starts every alignment record).
Remember -c says just count the records, don't display them. grep -P -c '^@' yeast_pairedend.sam
Or use the -v (invert) option to tell grep to print all lines that don't match a particular pattern, here the header lines starting with @. grep -P -v -c '^HWI' yeast_pairedend.sam
|
| Expand |
|---|
|
| There are 1184360 alignment records. |
Exercise: How many sequences were in the R1 and R2 FASTQ files combined?
| Expand |
|---|
|
gunzip -c fq/Sample_Yeast_L005_R1.cat.fastq.gz | echo $((`wc -l` / 2))
|
| Expand |
|---|
|
| There were a total of 1184360 original sequences |
Exercises:
- Do both R1 and R2 reads have separate alignment records?
- Does the SAM file contain both aligned and un-aligned reads?
- What is the order of the alignment records in this SAM file?
| Expand |
|---|
|
- Do both R1 and R2 reads have separate alignment records?
- yes, they must, because there were 1,184,360 R1+R2 reads and an equal number of alignment records
- Does the SAM file contain both aligned and un-aligned reads?
- yes, it must, because there were 1,184,360 R1+R2 reads and an equal number of alignment records
- What is the order of the alignment records in this SAM file?
- the names occur in the exact same order as they did in the FASTQ, except that they come in pairs
- the R1 read comes first, then its corresponding R2
- this ordering is called read name ordering
|
Exercise #2: Bowtie2 and Local Alignment - Human microRNA-seq
Now we're going to switch over to a different aligner that was originally designed for very short reads and is frequently used for RNA-seq data. Accordingly, we have prepared another test microRNA-seq dataset for you to experiment with (not the same one you used cutadapt on). This data is derived from a human H1 embryonic stem cell (H1-hESC) small RNA dataset generated by the ENCODE Consortium – its about a half million reads.
However, there is a problem! We don't know (or, well, you don't know) what the adapter structure or sequences were. So, you have a bunch of 36 base pair reads, but many of those reads will include extra sequence that can impede alignment – and we don't know where! We need an aligner that can find subsections of the read that do align, and discard (or "soft-clip") the rest away – an aligner with a local alignment mode. Bowtie2 is just such an aligner.
Overview miRNA alignment workflow with bowtie2
If the adapter structure were known, the normal workflow would be to first remove the adapter sequences with cutadapt. Since we can't do that, we will instead perform a local lignment of the single-end miRNA sequences using bowtie2. This workflow has the following steps:
- Prepare the mirbase v20 reference index for bowtie2 (one time) using bowtie2-build
- Perform local alignment of the R1 reads with bowtie2, producing a SAM file directly
- Convert the SAM file to a BAM file (samtools view)
- Sort the BAM file by genomic location (samtools sort)
- Index the BAM file (samtools index)
- Gather simple alignment statistics (samtools flagstat and samtools idxstat)
This looks so much simpler than bwa – only one alignment step instead of three! We'll see the price for this "simplicity" in a moment...
As before, we will just do the alignment steps leave samtools for the next section.
Introducing bowtie2
Go ahead and load the bowtie2 module so we can examine some help pages and options. To do that, you must first load the perl module, and then the a specific version of bowtie2.
| Code Block |
|---|
module load perl
module load bowtie/2.2.0
|
Now that it's loaded, check out the options. There are a lot of them! In fact for the full range of options and their meaning, Google "Bowtie2 manual" and bring up that page. Ouch!
This is the key to using bowtie2 - it allows you to control almost everything about its behavior, but that also makes it is much more challenging to use than bwa – and it's easier to screw things up too!
Building the bowtie2 mirbase index
Before the alignment, of course, we've got to build a mirbase index using bowtie2-build (go ahead and check out its options). Unlike for the aligner itself, we only need to worry about a few things here:
| Code Block |
|---|
bowtie2-build <reference_in> <bt2_index_base> |
Here, reference_in file is just the FASTA file containing mirbase v20 sequences, and bt2_index_base is the prefix of where we want the files to go. Following what we did earlier for BWA indexing:
| Code Block |
|---|
cd $SCRATCH/references/
mkdir mirbase
mv hairpin_cDNA_hsa.fa mirbase
cd mirbase
bowtie2-build hairpin_cDNA_hsa.fa hairpin_cDNA_hsa.fa |
That was very fast! It's because the mirbase reference genome is so small compared to what programs like this are used to dealing with, which is the human genome (or bigger). Now, your $SCRATCH/references/mirbase directory should be filled with the following files:
| Code Block |
|---|
hairpin_cDNA_hsa.fa
hairpin_cDNA_hsa.fa.1.bt2
hairpin_cDNA_hsa.fa.2.bt2
hairpin_cDNA_hsa.fa.3.bt2
hairpin_cDNA_hsa.fa.4.bt2
hairpin_cDNA_hsa.fa.rev.1.bt2
hairpin_cDNA_hsa.fa.rev.2.bt2 |
Now, we're ready to actually try to do the alignment. Remember, unlike BWA, we actually need to set some options depending on what we're after. These are the most important options when using Bowtie2:
| Option | Effect |
|---|
| -N | Controls the number of mismatches allowable in the seed of each alignment (default = 0) |
| -L | Controls the length of seed substrings generated from each read (default = 22) |
| --end-to-end or --local | Controls whether the entire read must align to the reference, or whether soft-clipping the ends is allowed to find internal alignments |
| -ma | Controls the alignment score contribution of a matching base (0 for --end-to-end, 2 for --local |
To decide how we want to go about doing our alignment, check out the file we're aligning with 'less'.
| Expand |
|---|
|
| Code Block |
|---|
cds
less fastq_align/human_mirnaseq.fastq.gz |
|
Lots of those reads have long strings of A's, which must be an adapter or protocol artifact. Even though we see how we might be able to fix it using some tools we've talked about, what if we had no idea what the adapter sequence was, or couldn't use cutadapt or other programs to prepare the reads? In that case, we need a local alignment where the seed length is the small boundary of the acceptable internal alignments. Here, we are interested in finding any sections of any reads that align well to a microRNA. These sequences are between 16 and 22 bases long, so any good alignment should have at least 16 matching bases, but could have more. Also, maybe we want to allow a mismatch or two in the seed, since we might be interested in miRNA SNPs. So, a good set of options might look something like this:
| Code Block |
|---|
-N 1 -L 16 --local |
This leaves the default scoring method as "-ma 2", meaning that a 16 base pair alignment will have a score of 32, and so on. This is VERY different from the alignment scores assigned by other aligners, so it's worth remembering.
As you can tell from looking at the Bowtie2 help message, the syntax looks like this:
| Code Block |
|---|
bowtie2 [options]* -x <bt2-idx> {-1 <m1> -2 <m2> | -U <r>} [-S <sam>] |
As such, our alignment command (now that we have the FASTQ file and the reference sequence ready) could be this (make sure you are located in your scratch directory!):
| Code Block |
|---|
cds
bowtie2 -N 1 -L 16 --local -x references/mirbase/hairpin_cDNA_hsa.fa -U fastq_align/human_mirnaseq.fastq.gz -S alignments/human_mirnaseq.sam |
Now, you should have a human_mirnaseq.sam file in your alignments directory, that you can check out using whatever commands you like. An example alignment looks like this:
| Code Block |
|---|
TUPAC_0037_FC62EE7AAXX:2:1:2607:1430#0/1 0 hsa-mir-302b 50 22 3S20M13S * 0 0 TACGTGCTTCCATGTTTTANTAGAAAAAAAAAAAAG ZZFQV]Z[\IacaWc]RZIBVGSHL_b[XQQcXQcc AS:i:37 XN:i:0 XM:i:1 XO:i:0 XG:i:0 NM:i:1 MD:Z:16G3 YT:Z:UU |
Note how the CIGAR string is 3S20M13S, meaning that 13 bases were soft clipped from one end, and 3 from the other. If we did the same alignment using either --end-to-end mode, or using BWA in the same way as we did in Exercise #1, very little of this file would have aligned. However, if we had not lowered the seed parameter of Bowtie2 from its default of 22, we would not have found many of the alignments like the one shown above, because the read only matched for 20 bases - a matching 22 base seed does not exist. Such is the nature of Bowtie2 - it can be a powerful tool to sift out the alignments you want from a messy dataset with limited information, but doing so requires careful tuning of the parameters, which can in itself take a lot of time to perfect.
Exercise #3: BWA-MEM (and Tophat2) - Human mRNA-seq
After Bowtie2 came out with a local alignment option, it wasn't long before BWA generated their own local-aligner called BWA-MEM (for Maximal Exact Matches). This aligner is very, very nice because it incorporates a lot of the simplicity of using BWA with the complexities of local alignment. This functionality, while enabling the alignment of datasets like the mirbase data we just examined, also permits more complex alignments, such as that of spliced mRNAs. In a long RNA-seq experiment, reads will (at some frequency) span a splice junction themselves, or a pair of reads in a paired-end library will fall on either side of a splice junction. We want to be able to align reads that do this for many reasons, from accurate transcript quantification to novel fusion transcript discovery. Thus, our last exercise will be the alignment of a human LONG RNA-seq dataset composed (by design) almost exclusively of reads that cross splice junctions.
BWA-MEM should have been loaded when we loaded the BWA module, so to look at the details of MEM alignment, just enter "bwa mem" to get the help menu with the options list. The most important parameters, similar to those we've manipulated in the past two sections, are the following:
| Option | Effect |
|---|
| -k | Controls the minimum seed length (default = 19) |
| -w | Controls the "gap bandwidth", or the length of a maximum gap. This is particularly relevant for MEM, since it can determine whether a read is split into two separate alignments, or one long alignment with a long gap in the middle (default = 100) |
| -r | Controls how long an alignment must be relative to its seed before it is re-seeded to try to find a best-fit local match (default = 1.5, e.g. the value of -k multiplied by 1.5) |
| -c | Controls how many matches a MEM must have in the genome before it is discarded (default = 10000) |
| -t | Controls the number of threads to use |
There are many more parameters to control the scoring scheme and other details, but these are the most essential parameters to use to get anything of value at all.
The test file we will be working with is JUST the R1 file from a paired-end total RNA-seq experiment, meaning it is (for our purposes) single-end. Go ahead and take a look at it, and find out how many reads are in the file.
| Expand |
|---|
|
| Code Block |
|---|
cds
less fastq_align/human_rnaaseq.fastq.gz
gunzip -c fastq_align/human_rnaseq.fastq.gz | echo $((`wc -l`/4)) |
|
Now, try aligning it with BWA like we did in example 1. This won't take very long, but you'll need to use our pre-indexed hg19 reference. It is located at:
| Code Block |
|---|
/scratch/01063/abattenh/ref_genome/bwa/bwtsw/hg19.fa |
if you look at the contents of the 'bwtsw' directory in the above path, you'll see a set of files that are analogous to the yeast files we created earlier, except that their universal prefix is 'hg19.fa'. Now, using that index, go ahead and try to do a single-end alignment of the file to the human genome like we did in Exercise #1, and save the contents to intermediate files with the prefix 'human_rnaseq_bwa'.
| Expand |
|---|
|
| Code Block |
|---|
cds
bwa aln /scratch/01063/abattenh/ref_genome/bwa/bwtsw/hg19/hg19.fa fastq_align/human_rnaseq.fastq.gz > alignments/human_rnaseq_bwa.sai
bwa samse /scratch/01063/abattenh/ref_genome/bwa/bwtsw/hg19/hg19.fa alignments/human_rnaseq_bwa.sai fastq_align/human_rnaseq.fastq.gz > alignments/human_rnaseq_bwa.sam |
|
Now, use less to take a look at the contents of the sam file, using the space bar to leaf through them. You'll notice a lot of alignments look basically like this:
| Code Block |
|---|
HWI-ST1097:228:C21WMACXX:8:1316:10989:88190 4 * 0 0 * * 0 0
AAATTGCTTCCTGTCCTCATCCTTCCTGTCAGCCATCTTCCTTCGTTTGATCTCAGGGAAGTTCAGGTCTTCCAGCCGCTCTTTGCCACTGATCTCCAGCT
CCCFFFFFHHHHHIJJJJIJJJJIJJJJHJJJJJJJJJJJJJJIIIJJJIGHHIJIJIJIJHBHIJJIIHIEGHIIHGFFDDEEEDDCDDD@CDEDDDCDD |
meaning they are not alignments at all. Essentially, nothing (with a few exceptions) aligned. That's because this file was generated exclusively from reads in a larger dataset that cross at least one splice junction (surprise!), meaning that they sequence as it exists in most of the reads does not exist anywhere in the genome, but some subsections of each read do exist somewhere in the genome. So, we need an aligner that is capable of finding regions of each read (above some length cutoff) that align to the genome. Based on the following syntax, and using the reference path, we noted earlier, try to use BWA MEM to align the same file, single-end, saving all output files with the prefix 'human_rnaseq_mem':
| Code Block |
|---|
bwa mem ref.fa reads.fq > aln-se.sam |
| Expand |
|---|
|
| Code Block |
|---|
bwa mem /scratch/01063/abattenh/ref_genome/bwa/bwtsw/hg19/hg19.fa fastq_align/human_rnaseq.fastq.gz > alignments/human_rnaseq_mem.sam |
|
Now, check the length of the SAM file you generated with 'wc -l'. Since there is one alignment per line, there must be 586266 alignments, which is more than the number of sequences in the FASTQ file. This is because many reads will align twice or more, hopefully on either side of a splice junction. These alignments can still be associated because they will have the same read ID, but are reflected in more than one line. To get an idea of how often each read aligned, and what the 'real' alignment rate is, use the following commands:
| Code Block |
|---|
cut -f 1 alignments/human_rnaseq_mem.sam | sort | uniq -c | less #This gives you a view where each read is listed next to the number of entries it has in the SAM file
cut -f 1 alignments/human_rnaseq.sam | sort | uniq -c | awk '{print $1}' | sort | uniq -c | less #This gives essentially a histogram of the number of times each read aligned - a plurality of reads aligned twice, which seems reasonable since these are all reads crossing a junction, but plenty aligned more or less
cut -f 1 alignments/human_rnaseq.sam | sort | uniq | wc -l #This gives a better idea of the alignment rate, which is how many reads aligned at least once to the genome. Divided by the number of reads in the original file, the real alignment rate is around 64.19 %.
# PLEASE NOTE: some of these one-liners are only reasonably fast if the files are relatively small (around a million reads or less). For bigger files, there are better ways to get this information, mostly using samtools, which is a utility explicitly designed for manipulating SAM/BAM files. We'll cover samtools in the next section. |
If you want to be able to generate a file with only the best (for BWA MEM, that means the longest) alignment for each read, so that every read can only be represented by one line, the option -M will add a flag to the end of each alignment entry that designates any alignments that are not the longest for their originating read as secondary. Then, you can filter all reads that do not have that flag.
...