Overview
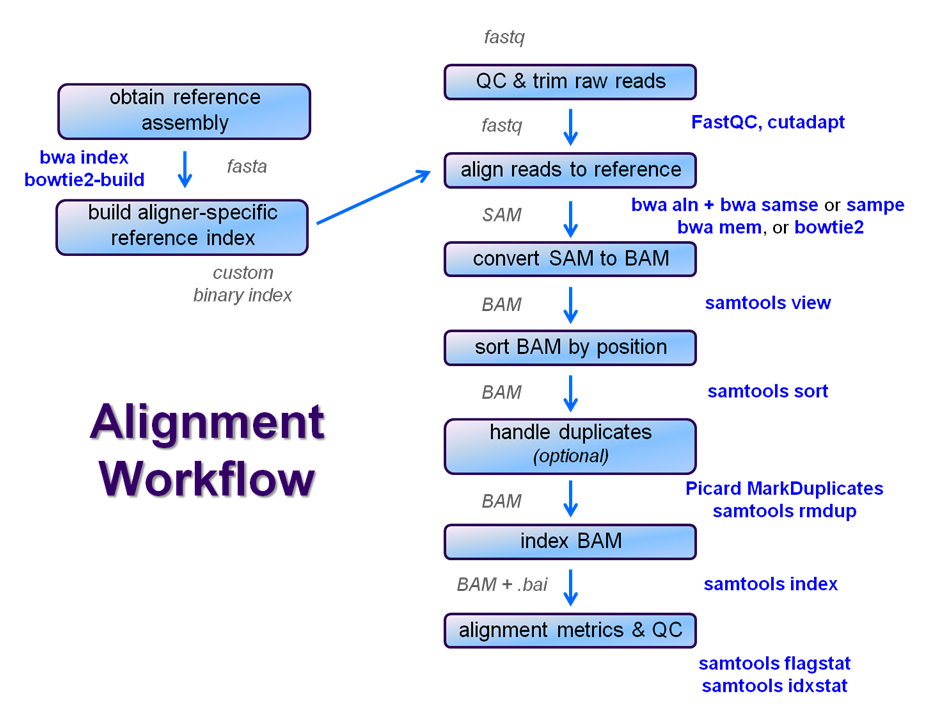
After raw sequence files are generated (in FASTQ format), quality-checked, and pre-processed in some way, the next step in most NGS pipelines is mapping to a reference genome.
For individual sequences, it is common to use a tool like BLAST to identify genes or species of origin. However, a normal NGS dataset will have tens to hundreds of millions of sequences, which BLAST and similar tools are not designed to handle. Thus, a large set of computational tools have been developed to quickly, align each read to its best location, if any, in a reference.
Even though many mapping tools exist, a few individual programs have a dominant "market share" of the NGS world. In this section, we will primarily focus on two of the most versatile mappers: BWA and Bowtie2, the latter being part of the Tuxedo suite which includes the transcriptome-aware RNA-seq aligner Tophat2 as well as other downstream quantifiaction tools.
Connect to login5.ls5.tacc.utexas.edu
This should be second nature by now ![]()
Sample Datasets
You have already worked with a paired-end yeast ChIP-seq dataset, which we will continue to use here. For the sake of uniformity, however, we will set up a new directory in your scratch area called 'alignment' and fill it with our sequencing data. To set up your scratch area properly and move into it, execute something like:
mkdir -p $SCRATCH/core_ngs/alignment cd $SCRATCH/core_ngs/alignment mkdir fastq
Now you have created the alignment directory, moved into it, and created a subdirectory for our raw fastq files. We will be using four data sets that consist of five files (since the paired-end data set has two separate files for each of the R1 and R2 reads). To copy them over, execute something like:
cd $SCRATCH/core_ngs/alignment/fastq cp /corral-repl/utexas/BioITeam/core_ngs_tools/alignment/*fastq.gz .
We first moved ourselves into the fastq directory, then copied over all files that end in "fastq.gz" in the corral directory specified in the second line. These are descriptions of the five files we copied:
| File Name | Description | Sample |
|---|---|---|
| Sample_Yeast_L005_R1.cat.fastq.gz | Paired-end Illumina, First of pair, FASTQ | Yeast ChIP-seq |
| Sample_Yeast_L005_R2.cat.fastq.gz | Paired-end Illumina, Second of pair, FASTQ | Yeast ChIP-seq |
| human_rnaseq.fastq.gz | Paired-end Illumina, First of pair only, FASTQ | Human RNA-seq |
| human_mirnaseq.fastq.gz | Single-end Illumina, FASTQ | Human microRNA-seq |
| cholera_rnaseq.fastq.gz | Single-end Illumina, FASTQ | V. cholerae RNA-seq |
Reference Genomes
Before we get to alignment, we need a genome to align to. We will use four different references here:
- the yeast genome (sacCer3)
- the human genome (hg19)
- a Vibrio cholerae genome (0395; our name: vibCho)
- the microRNA database mirbase (v20), human subset
NOTE: For the sake of simplicity, these are not necessarily the most recent versions of these references - for example, hg19 is the second most recent human genome, with the most recent called hg38. Similarly, the most recent mirbase annotation is v21.
These are the four reference genomes we will be using today, with some information about them (and here is information about many more genomes):
| Reference | Species | Base Length | Contig Number | Source | Download |
|---|---|---|---|---|---|
| hg19 | Human | 3.1 Gbp | 25 (really 93) | UCSC | UCSC GoldenPath |
| sacCer3 | Yeast | 12.2 Mbp | 17 | UCSC | UCSC GoldenPath |
| mirbase V20 | Human | 160 Kbp | 1908 | Mirbase | Mirbase Downloads |
| vibCho (O395) | V. cholerae | ~4 Mbp | 2 | GenBank | GenBank Downloads |
Searching genomes is computationally hard work and takes a long time if done on un-indexed, linear genomic sequence. So aligners require that references first be indexed to accelerate later retrieval. The aligners we are using each require a different index, but use the same method (the Burrows-Wheeler Transform) to get the job done. This involves taking a FASTA file as input, with each chromosome (or contig) as a separate FASTA entry, and producing an aligner-specific set of files as output. Those output index files are then used by the aligner when performing the sequence alignment, and subsequent alignments are reported using coordinates referencing the original FASTA reference files.
hg19 is way too big for us to index here, so we're not going to do it (especially not all at the same time!). Instead, we will "point" to an existing set of hg19 index files, which are all located at:
/scratch/01063/abattenh/ref_genome/bwa/bwtsw/hg19
We can index the references for the yeast genome, the human miRNAs, and the V. cholerae genome, because they are all tiny compared to the human genome. We will grab the FASTA files for yeast and human miRNAs two references and build each index right before we use them. We will also grab the special file that contains the V. cholerae genome sequence and annotations (a .gbk file), and generate the reference FASTA and some other interesting information when we get to that exercise. These references are currently at the following locations:
/corral-repl/utexas/BioITeam/core_ngs_tools/references/sacCer3.fa /corral-repl/utexas/BioITeam/core_ngs_tools/references/hairpin_cDNA_hsa.fa /corral-repl/utexas/BioITeam/core_ngs_tools/references/vibCho.O395.gbk
First stage all the reference files in your $WORK core_ngs area in a directory called references. We will add further structure to this directory later on in specific exercises, but for now the following will suffice:
mkdir -p $WORK/core_ngs/references cp $CLASSDIR/references/* $WORK/core_ngs/references $CLASSDIR = /corral-repl/utexas/BioITeam/core_ngs_tools
With that, we're ready to get started on the first exercise.
Exercise #1: BWA global alignment – Yeast ChIP-seq
Overview ChIP-seq alignment workflow with BWA
We will perform a global alignment of the paired-end Yeast ChIP-seq sequences using bwa. This workflow generally has the following steps:
- Trim the FASTQ sequences down to 50 with fastx_clipper
- this removes most of any 5' adapter contamination without the fuss of specific adapter trimming w/cutadapt
- Prepare the sacCer3 reference index for bwa using bwa index
- this is done once, and re-used for later alignments
- Perform a global bwa alignment on the R1 reads (bwa aln) producing a BWA-specific binary .sai intermediate file
- Perform a global bwa alignment on the R2 reads (bwa aln) producing a BWA-specific binary .sai intermediate file
- Perform pairing of the separately aligned reads and report the alignments in SAM format using bwa sampe
- Convert the SAM file to a BAM file (samtools view)
- Sort the BAM file by genomic location (samtools sort)
- Index the BAM file (samtools index)
- Gather simple alignment statistics (samtools flagstat and samtools idxstat)
We're going to skip the trimming step for now and see how it goes. We'll perform steps 2 - 5 now and leave samtools for the next course section, since those steps (6 - 10) are common to nearly all post-alignment workflows.
Introducing BWA
Like other tools you've worked with so far, you first need to load bwa using the module system. Go ahead and do that now, and then enter bwa with no arguments to view the top-level help page (many NGS tools will provide some help when called with no arguments).
module load bwa bwa
As you can see, bwa include many sub-commands that perform most of the tasks we are interested in.
Building the BWA sacCer3 index
We're going to index the genome with the index command. To learn what this sub-command needs in the way of options and arguments, enter bwa index with no arguments.
Usage: bwa index [-a bwtsw|is] [-c] <in.fasta>
Options: -a STR BWT construction algorithm: bwtsw or is [auto]
-p STR prefix of the index [same as fasta name]
-b INT block size for the bwtsw algorithm (effective with -a bwtsw) [10000000]
-6 index files named as <in.fasta>.64.* instead of <in.fasta>.*
Warning: `-a bwtsw' does not work for short genomes, while `-a is' and
`-a div' do not work not for long genomes. Please choose `-a'
according to the length of the genome.
Based on the "Usage" description, we only need to specify two things:
- the name of the FASTA file
- whether to use the bwtsw or is algorithm for indexing
Since sacCer3 is relative large (~12 Mbp) we will specify bwtsw as the indexing option (as indicated by the "Warning" message), and the name of the FASTA file is sacCer3.fa.
Importantly, the output of this command is a group of files that are all required together as the index. So, within the references directory, we will create another directory called bwa/sacCer3, make a symbolic link to the yeast FASTA there, and run the index command in that directory. So, first we will create a directory called fasta to hold our reference FASTA file, then create the bwa/sacCer3 directory, and construct the symbolic links.
mkdir -p $WORK/core_ngs/references/bwa/sacCer3 mkdir -p $WORK/core_ngs/references/fasta mv $WORK/core_ngs/references/sacCer3.fa $WORK/core_ngs/references/fasta cd $WORK/core_ngs/references/bwa/sacCer3 ln -s ../../fasta/sacCer3.fa ls -la
Now execute the bwa index command.
bwa index -a bwtsw sacCer3.fa
Since the yeast genome is not large when compared to human, this should not take long to execute (otherwise we would do it as a batch job). When it is complete you should see a set of index files like this:
sacCer3.fa sacCer3.fa.amb sacCer3.fa.ann sacCer3.fa.bwt sacCer3.fa.pac sacCer3.fa.sa
Exploring the FASTA with grep
It is frequently useful to have a list of all contigs/chromosomes/genes/features in a file. You'll usually want to know this before you start the alignment so that you're familiar with the contig naming convention – and to verify that it's the one you expect. For example, chromosome 1 is specified as "chr1", "1", "I", and more in different references, and it can get weird for non-model organisms.
We saw that a FASTA consists of a number of contig entries, each one starting with a name line of the form below, followed by many lines of bases.
>contigName
How do we dig out just the lines that have the contig names and ignore all the sequences? Well, the contig name lines all follow the pattern above, and since the > character is not a valid base, it will never appear on a sequence line.
We've discovered a pattern (also known as a regular expression) to use in searching, and the command line tool that does regular expression matching is grep.
Regular expressions are so powerful that nearly every modern computer language includes a "regex" module of some sort. There are many online tutorials for regular expressions (and a few different flavors of them). But the most common is the Perl style (http://perldoc.perl.org/perlretut.html). We're only going to use the most simple of regular expressions here, but learning more about them will pay handsome dividends for you in the future (there's a reason Perl was used a lot when assembling the human genome).
Here's how to execute grep to list contig names in a FASTA file.
grep -P '^>' sacCer3.fa | more
Notes:
- The -P option tells grep to use Perl-style regular expression patterns.
- This makes including special characters like Tab ( \t ), Carriage Return ( \r ) or Linefeed ( \n ) much easier that the default Posix paterns.
- While it is not really required here, it generally doesn't hurt to include this option.
'^>' is the regular expression describing the pattern we're looking for (described below)
- sacCer3.fa is the file to search. Lines with text that match our pattern will be written to standard output; non matching lines will be omitted.
- We pipe to more just in case there are a lot of contig names.
Now down to the nuts and bolts of our pattern, '^>'
First, the single quotes around the pattern – they are only a signal for the bash shell. As part of its friendly command line parsing and evaluation, the shell will often look for special characters on the command line that mean something to it (for example, the $ in front of an environment variable name, like in $SCRATCH). Well, regular expressions treat the $ specially too – but in a completely different way! Those single quotes tell the shell "don't look inside here for special characters – treat this as a literal string and pass it to the program". The shell will obey, will strip the single quotes off the string, and will pass the actual pattern, ^>, to the grep program. (Aside: We've see that the shell does look inside double quotes ( " ) for certain special signals, such as looking for environment variable names to evaluate.)
So what does ^> mean to grep? Well, from our contig name format description above we see that contig name lines always start with a > character, so > is a literal for grep to use in its pattern match.
We might be able to get away with just using this literal alone as our regex, specifying '>' as the command line argument. But for grep, the more specific the pattern, the better. So we constrain where the > can appear on the line. The special carat ( ^ ) character represents "beginning of line". So ^> means "beginning of a line followed by a > character, followed by anything. (Aside: the dollar sign ( $ ) character represents "end of line" in a regex. There are many other special characters, including period ( . ), question mark ( ? ), pipe ( | ), parentheses ( ( ) ), and brackets ( [ ] ), to name the most common.)
Exercise: How many contigs are there in the sacCer3 reference?
Performing the bwa alignment
Now, we're ready to execute the actual alignment, with the goal of initially producing a SAM file from the input FASTQ files and reference. First go to the align directory, and link to the sacCer3 reference directory (this will make our commands more readable).
cd $SCRATCH/core_ngs/alignment ln -s $WORK/core_ngs/references/bwa/sacCer3 ls
As our workflow indicated, we first use bwa aln on the R1 and R2 FASTQs, producing a BWA-specific .sai intermediate binary files. Since these alignments are completely independent, we can execute them in parallel in a batch job.
What does bwa aln needs in the way of arguments?
There are lots of options, but here is a summary of the most important ones. BWA, is a lot more complex than the options let on. If you look at the BWA manual on the web for the aln sub-command, you'll see numerous options that can increase the alignment rate (as well as decrease it), and all sorts of other things.
| Option | Effect |
|---|---|
| -l | Controls the length of the seed (default = 32) |
| -k | Controls the number of mismatches allowable in the seed of each alignment (default = 2) |
| -n | Controls the number of mismatches (or fraction of bases in a given alignment that can be mismatches) in the entire alignment (including the seed) (default = 0.04) |
| -t | Controls the number of threads |
The rest of the options control the details of how much a mismatch or gap is penalized, limits on the number of acceptable hits per read, and so on. Much more information can be accessed at the BWA manual page.
For a simple alignment like this, we can just go with the default alignment parameters.
Also note that bwa writes its (binary) output to standard output by default, so we need to redirect that to a .sai file.
For simplicity, we will just execute these commands directly, one at a time. (We can do this since we're on the special login8 head node!) Each command you execute should only take a minute or so.
bwa aln sacCer3/sacCer3.fa fastq/Sample_Yeast_L005_R1.cat.fastq.gz > yeast_R1.sai bwa aln sacCer3/sacCer3.fa fastq/Sample_Yeast_L005_R2.cat.fastq.gz > yeast_R2.sai
When all is done you should have two .sai files.
Next we use the bwa sampe command to pair the reads and output SAM format data. Just type that command in with no arguments to see its usage.
For this command you provide the same reference index prefix as for bwa aln, along with the two .sai files and the two original FASTQ files. Also, bwa writes its output to standard output, so redirect that to a .sam file.
Here is the command line statement you need. Just execute it on the command line.
bwa sampe sacCer3/sacCer3.fa yeast_R1.sai yeast_R2.sai fastq/Sample_Yeast_L005_R1.cat.fastq.gz fastq/Sample_Yeast_L005_R2.cat.fastq.gz > yeast_pairedend.sam
You did it! You should now have a SAM file that contains the alignments. It's just a text file, so take a look with head, more, less, tail, or whatever you feel like. In the next section, with samtools, you'll learn some additional ways to analyze the data once you create a BAM file.
Exercise: What kind of information is in the first lines of the SAM file?
Exercise: How many alignment records (not header records) are in the SAM file?
Exercise: How many sequences were in the R1 and R2 FASTQ files combined?
Exercises:
- Do both R1 and R2 reads have separate alignment records?
- Does the SAM file contain both aligned and un-aligned reads?
- What is the order of the alignment records in this SAM file?
Using cut to isolate fields
Recall the format of a SAM/BAM file alignment record:

Suppose you wanted to look only at field 3 (contig name) values in the SAM file. You can do this with the handy cut command. Below is a simple example where you're asking cut to display the 3rd column value for the last 10 alignment records.
tail yeast_pairedend.sam | cut -f 3
By default cut assumes the field delimiter is Tab, which is the delimiter used in the majority of NGS file formats. You can, of course, specify a different delimiter with the -d option.
You can also specify a range of fields, and mix adjacent and non-adjacent fields. This displays fields 2 through 6, field 9, and all fields starting with 12 (SAM tag fields).
tail yeast_pairedend.sam | cut -f 2-6,9,12-
You may have noticed that some alignment records contain contig names (e.g. chrV) in field 3 while others contain an asterisk ( * ). Usually the * means the record didn't align. (This isn't always true – later you'll see how to properly distinguish between mapped and unmapped reads using samtools.) We're going to use this heuristic along with cut to see about how many records represent aligned sequences.
First we need to make sure that we don't look at fields in the SAM header lines. We're going to end up with a series of pipe operations, and the best way to make sure you're on track is to enter them one at a time piping to head:
# the ^HWI pattern matches lines starting with HWI (the start of all read names in column 1) grep -P '^HWI' yeast_pairedend.sam | head
Ok, it looks like we're seeing only alignment records. Now let's pull out only field 3 using cut:
grep -P '^HWI' yeast_pairedend.sam | cut -f 3 | head
Cool, we're only seeing the contig name info now. Next we use grep again, piping it our contig info and using the -v (invert) switch to say print lines that don't match the pattern:
grep -P '^HWI' yeast_pairedend.sam | cut -f 3 | grep -v '*' | head
Perfect! We're only seeing real contig names that (usually) represent aligned reads. Let's count them by piping to wc -l (and omitting omit head of course – we want to count everything).
grep -P '^HWI' yeast_pairedend.sam | cut -f 3 | grep -v '*' | wc -l
Exercise: About how many records represent aligned sequences? What alignment rate does this represent?
Exercise: What might we try in order to improve the alignment rate?
Exercise #5: Simple SAMtools Utilities
We have used several alignment methods that all generate results in the form of the near-universal SAM/BAM file format. The SAMtools program is a ubiquitously used set of tools that allow a user to manipulate SAM/BAM files in many different ways, ranging from simple tasks (like SAM/BAM interconversion) to more complex functions (like removal of PCR duplicates). It is available in the TACC module system in the typical fashion.
In this exercise, we will use five very simple utilities provided by samtools: view, sort, index, flagstat, and idxstats. Each of these is executed in one line for a given SAM/BAM file. In the SAMtools/BEDtools section tomorrow we will explore samtools in more in depth.
For the sake of time and simplicity, here we are only going to run these commands on the yeast paired-end alignment file. The same commands can be run on the other files by changing the names, so feel free to try them on other SAM files. Indeed, it is very common in practice to use bash loops to generate many commands for a large set of alignments and deposit those commands into a batch job cmds file for submission.
To start, we will move to the directory containing our SAM files, among other things, and load up samtools using the module system. After loading it, just run the samtools command to see what the available tools are (and to see what the syntax of an actual command is).
cd $SCRATCH/core_ngs/alignment ls -la module load samtools samtools
You will see the following screen after running samtools with no other options:
Program: samtools (Tools for alignments in the SAM format)
Version: 1.2 (using htslib 1.2.1)
Usage: samtools <command> [options]
Commands:
-- indexing
faidx index/extract FASTA
index index alignment
-- editing
calmd recalculate MD/NM tags and '=' bases
fixmate fix mate information
reheader replace BAM header
rmdup remove PCR duplicates
targetcut cut fosmid regions (for fosmid pool only)
-- file operations
bamshuf shuffle and group alignments by name
cat concatenate BAMs
merge merge sorted alignments
mpileup multi-way pileup
sort sort alignment file
split splits a file by read group
bam2fq converts a BAM to a FASTQ
-- stats
bedcov read depth per BED region
depth compute the depth
flagstat simple stats
idxstats BAM index stats
phase phase heterozygotes
stats generate stats (former bamcheck)
-- viewing
flags explain BAM flags
tview text alignment viewer
view SAM<->BAM<->CRAM conversion
SAMtools version differences
Be sure to check what version of samtools you are using!
The most recent edition of SAMtools is 1.2, which has some important differences from the last version, 0.1.19. Most commands for this section are the same between the two versions, but if you see code from other sources using samtools, the version differences may be important.
Samtools view
The utility samtools view provides a way of converting SAM (text format) files to BAM (binary, compressed) files directly. It also provides many, many other functions which we will discuss lster. To get a preview, execute samtools view without any other arguments. You should see:
Usage: samtools view [options] <in.bam>|<in.sam>|<in.cram> [region ...]
Options: -b output BAM
-C output CRAM (requires -T)
-1 use fast BAM compression (implies -b)
-u uncompressed BAM output (implies -b)
-h include header in SAM output
-H print SAM header only (no alignments)
-c print only the count of matching records
-o FILE output file name [stdout]
-U FILE output reads not selected by filters to FILE [null]
-t FILE FILE listing reference names and lengths (see long help) [null]
-T FILE reference sequence FASTA FILE [null]
-L FILE only include reads overlapping this BED FILE [null]
-r STR only include reads in read group STR [null]
-R FILE only include reads with read group listed in FILE [null]
-q INT only include reads with mapping quality >= INT [0]
-l STR only include reads in library STR [null]
-m INT only include reads with number of CIGAR operations
consuming query sequence >= INT [0]
-f INT only include reads with all bits set in INT set in FLAG [0]
-F INT only include reads with none of the bits set in INT
set in FLAG [0]
-x STR read tag to strip (repeatable) [null]
-B collapse the backward CIGAR operation
-s FLOAT integer part sets seed of random number generator [0];
rest sets fraction of templates to subsample [no subsampling]
-@ INT number of BAM compression threads [0]
-? print long help, including note about region specification
-S ignored (input format is auto-detected)
That is a lot to process! For now, we just want to read in a SAM file and output a BAM file. The input format is auto-detected, so we don't need to say that we're inputing a SAM instead of a BAM. We just need to tell the tool to output the file in BAM format, and provide the name of the destination BAM file. This command is as follows:
samtools view -b yeast_pairedend.sam -o yeast_pairedend.bam
- the -b option tells the tool to output BAM format
- the -o option specifies the name of the output BAM file that will be created
How do you look at the BAM file contents now? That's simple. Just use samtools view. without the -b option. Remember to pipe output to a pager!
samtools view yeast_pairedend.bam | more
Samtools sort
Look at the SAM file briefly using less. You will notice, if you scroll down, that the alignments are in no particular order, with chromosomes and start positions all mixed up. This makes searching through the file very inefficient. samtools sort is a piece of samtools that provides the ability to re-order entries in the SAM file either by coordinate position or by read name.
If you execute samtools sort without any options, you see its help page:
Usage: samtools sort [options...] [in.bam]
Options:
-l INT Set compression level, from 0 (uncompressed) to 9 (best)
-m INT Set maximum memory per thread; suffix K/M/G recognized [768M]
-n Sort by read name
-o FILE Write final output to FILE rather than standard output
-O FORMAT Write output as FORMAT ('sam'/'bam'/'cram') (either -O or
-T PREFIX Write temporary files to PREFIX.nnnn.bam -T is required)
-@ INT Set number of sorting and compression threads [1]
Legacy usage: samtools sort [options...] <in.bam> <out.prefix>
Options:
-f Use <out.prefix> as full final filename rather than prefix
-o Write final output to stdout rather than <out.prefix>.bam
-l,m,n,@ Similar to corresponding options above
In most cases you will be sorting a BAM file by position rather than by name. You can use either -o or reidrection with > to control the output.
To sort the paired-end yeast BAM file by coordinate, and get a BAM file named yeast_pairedend.sort.bam as output, execute the following command:
samtools sort -O bam -T yeast_pairedend.sort yeast_pairedend.bam > yeast_pairedend.sort.bam
- The -O options says the output format should be BAM
- The -T options gives a prefix for temporary files produced during sorting
- By default sort writes its output to standard output, so we use > to redirect to a file named yeast_pairedend.sort.bam
Samtools index
Many tools (like the UCSC Genome Browser) only need to use sub-sections of the BAM file at a given point in time. For example, if you are viewing alignments that are within a particular gene alignments on other chromosomes generally do not need to be loaded. In order to speed up access, BAM files are indexed, producing BAI files which allow other programs to navigate directly to the alignments of interest. This is especially important when you have many alignments.
The utility samtools index creates an index that has the exact name as the input BAM file, with suffix .bai appended. The help page, if you execute samtools index with no arguments, is as follows:
Usage: samtools index [-bc] [-m INT] <in.bam> [out.index] Options: -b Generate BAI-format index for BAM files [default] -c Generate CSI-format index for BAM files -m INT Set minimum interval size for CSI indices to 2^INT [14]
Here, the syntax is way, way easier. We want a BAI-format index which is the default. (CSI-format is used with extremely long contigs, which we aren't considering here - the most common use case are highly polyploid plant genomes).
So all we have to type is:
samtools index yeast_pairedend.sort.bam
This will produce a file named yeast_pairedend.bam.bai.
Most of the time when an index is required, it will be automatically located as long as it is in the same directory as its BAM file and shares the same name up until the .bai extension.
Samtools idxstats
Now that we have a sorted, indexed BAM file, we might like to get some simple statistics about the alignment run. For example, we might like to know how many reads aligned to each chromosome/contig. The samtools idxstats is a very simple tool that provides this information. If you type the command without any arguments, you will see that it could not be simpler - just type the following command:
samtools idxstats yeast_pairedend.sort.bam
The output is a text file with four tab-delimited columns with the following meanings:
- chromosome name
- chromosome length
- number of mapped reads
- number of unmapped reads
The reason that the "unmapped reads" field for the named chromosomes is not zero is that, if one half of a pair of reads aligns while the other half does not, the unmapped read is still assigned to the chromosome its mate mapped to, but is flagged as unmapped.
If you're mapping to a non-genomic reference such as miRBase miRNAs or another set of genes (a transcriptome), samtools idxstats gives you a quick look at quantitative alignment results.
Samtools flagstat
Finally, we might like to obtain some other statistics, such as the percent of all reads that aligned to the genome. The samtools flagstat tool provides very simple analysis of the SAM flag fields, which includes information like whether reads are properly paired, aligned or not, and a few other things. Its syntax is identical to that of samtools idxstats:
samtools flagstat yeast_pairedend.sort.bam
You should see something like this:
1184360 + 0 in total (QC-passed reads + QC-failed reads) 0 + 0 secondary 0 + 0 supplementary 0 + 0 duplicates 547664 + 0 mapped (46.24%:-nan%) 1184360 + 0 paired in sequencing 592180 + 0 read1 592180 + 0 read2 473114 + 0 properly paired (39.95%:-nan%) 482360 + 0 with itself and mate mapped 65304 + 0 singletons (5.51%:-nan%) 534 + 0 with mate mapped to a different chr 227 + 0 with mate mapped to a different chr (mapQ>=5)
Ignore the "+ 0" addition to each line - that is a carry-over convention for counting QA-failed reads that is no longer necessary.
The most important statistic is the mapping rate, but this readout allows you to verify that some common expectations (e.g. that about the same number of R1 and R2 reads aligned, and that most mapped reads are proper pairs) are met.
Exercise #6: Yeast BWA PE alignment with Anna's script
Now that you've done everything the hard way, let's see how to do run an alignment pipeline using Anna's script.
First the setup:
mkdir -p $SCRATCH/core_ngs/align2/fastq cd $SCRATCH/core_ngs/align2/fastq cp /corral-repl/utexas/BioITeam/core_ngs_tools/alignment/*fastq.gz .
Before executing the script you need to have one environment variable set. We'll do it at the command line here, but you could put it in your .bashrc file.
export path_code=/work/01063/abattenh/code
Now change into the directory and call the script with no arguments to see usage
cd $SCRATCH/core_ngs/align2 $path_code/script/align/align_bwa_illumina.sh
There are lots of bells and whistles in the arguments, but the most important are the first few:
- FASTQ file – full or relative path to the FASTQ file (just the R1 fastq if paired end). Can be compressed (.gz)
- output prefix – prefix for all the output files produced by the script. Should relate back to what the data is.
- assembly – genome assembly to use.
- there are pre-built indexes for some common eukaryotes (hg19, hg18, mm10, mm9, danRer7, sacCer3) that you can use
- or provide a full path for a bwa reference index you have built somewhere
- paired flag – 0 means single end (the default); 1 means paired end
- hard trim length – if you want the FASTQ hard trimmed down to a specific length, supply that number here
Now run the pipeline. By piping the output to tee <filename> we can see the script's progress at the terminal, and it also is written to <filename>.
$path_code/script/align/align_bwa_illumina.sh ./fastq/Sample_Yeast_L005_R1.cat.fastq.gz yeast_chip sacCer3 1 2>&1 | tee aln.yeast_chip.log
Output files
This alignment pipeline script performs the following steps:
- Hard trims FASTQ, if optionally specified (fastx_trimmer)
- Aligns the R1 FASTQ (bwa aln)
- Aligns the R2 FASTQ, if paired end alignment specified (bwa aln)
- Reports the alignments as SAM (bwa samse for single end, or bwa sampe for paired end)
- Converts SAM to BAM (samtools view)
- Sorts the BAM (samtools sort)
- Marks duplicates (Picard MarkDuplicates)
- Indexes the sorted, duplicate-marked BAM (samtools index)
- Gathers statistics (samtools idxstats, samtools flagstat, plus a custom statistics script of Anna's)
- Removes intermediate files
There are a number of output files, with the most important being those desribed below.
- aln.<prefix>.log – Log file of the entire alignment process.
- check the tail of this file to make sure the alignment was successful
- <prefix>.sort.dup.bam – Sorted, duplicate-marked alignment file.
- <prefix>.sort.dup.bam.bai – Index for the sorted, duplicate-marked alignment file
- <prefix>.samstats.txt – Summary alignment statistics from Anna's stats script
Verifying alignment success
The alignment log will have a "I ran successfully" message at the end if all went well, and if there was an error, the important information should also be at the end of the log file. So you can use tail to check the status of an alignment; for example:
tail aln.yeast_chip.log
This will show something like:
..samstats file 'yeast_chip.samstats.txt' exists Thu May 28 16:36:01 CDT 2015 ..samstats file file 'yeast_chip.samstats.txt' size ok Thu May 28 16:36:01 CDT 2015 --------------------------------------------------------- Cleaning up files... --------------------------------------------------------- ckRes 0 cleanup --------------------------------------------------------- All bwa alignment tasks completed successfully! Thu May 28 16:36:01 CDT 2015 ---------------------------------------------------------
Notice that success message: "All bwa alignment tasks completed successfully!". It should only appear once in any successful alignment log.
When multiple alignment commands are run in parallel it is important to check them all, and you can use grep looking for part of the unique success message to do this.
For example, suppose I have run 6 alignments and have these 6 log files:
aln.delswr1_htz1_tap1t0.log aln.delswr1_htz1_tap1t30.log aln.wt_htz1_tap1t15.log aln.delswr1_htz1_tap1t15.log aln.wt_htz1_tap1t0.log aln.wt_htz1_tap1t30.log
I can check that all 6 completed with this command:
grep 'completed successfully' aln.*.log | wc -l
If this command returns 6, I'm done. But what if it doesn't? If you grep -v (lines that don't contain the pattern), you'll get every line in every log file except the success message line, which is not what you want at all.
You could tail the log files one by one to see which one(s) don't have the message, but you can also use a special grep option to do this work:
grep -L 'completed successfully' aln.*.log
The -L option tells grep to only print the filenames that don't contain the pattern. Perfect!
Checking alignment statistics
The <prefix>.samstats.txt statistics file produced by the alignment pipeline has a lot of good information in one place. If you use cat or more to view it you'll see this:
-----------------------------------------------
Aligner: bwa
Total sequences: 1184360
Total mapped: 547664 (46.2 %)
Total unmapped: 636696 (53.8 %)
Primary: 547664 (100.0 %)
Secondary:
Duplicates: 324280 (59.2 %)
Fwd strand: 272898 (49.8 %)
Rev strand: 274766 (50.2 %)
Multi hit: 18688 (3.4 %)
Soft clip: 222451 (40.6 %)
All match: 319429 (58.3 %)
Indels: 6697 (1.2 %)
Spliced:
-----------------------------------------------
Total PE seqs: 1184360
PE seqs mapped: 547664 (46.2 %)
Num PE pairs: 592180
F5 1st end mapped: 300477 (50.7 %)
F3 2nd end mapped: 247187 (41.7 %)
PE pairs mapped: 241180 (40.7 %)
PE proper pairs: 236557 (39.9 %)
-----------------------------------------------
Insert size stats for: yeast_chip
Number of pairs: 236557 (proper)
Number of insert sizes: 212
Mean [-/+ 1 SD]: 215 [153 277] (sd 62)
Mode [Fivenum]: 223 [105 210 220 229 321]
-----------------------------------------------
Since this was a paired end alignment there is paired-end specific information reported, including insert size statistics: mean/standard deviation, mode (most common insert size value) and fivenum (min, q1, median, q3 max insert sizes).
A quick way to check alignment stats if you have run multiple alignments is again to use grep. For example, for the 6 alignment files shown earlier, running this:
grep 'Total map' *samstats.txt
will produce output like this:
delswr1_htz1_tap1t0.samstats.txt: Total mapped: 32761761 (86.8 %) delswr1_htz1_tap1t15.samstats.txt: Total mapped: 33699464 (89.2 %) delswr1_htz1_tap1t30.samstats.txt: Total mapped: 28441655 (87.6 %) wt_htz1_tap1t0.samstats.txt: Total mapped: 28454847 (89.5 %) wt_htz1_tap1t15.samstats.txt: Total mapped: 33245627 (90.9 %) wt_htz1_tap1t30.samstats.txt: Total mapped: 32567026 (90.7 %)
TACC batch system considerations
The great thing about pipeline scripts like this is that you can perform alignments on many datasets in parallel at TACC.
Anna's alignment pipeline scripts are written to take advantage of having multiple cores on TACC nodes, and are thus designed to run with at most two pipeline commands per TACC node.
Always specify wayness 2 for these pipeline scripts
These pipeline scripts should always be run with a wayness of 2 (-w 2) in the TACC batch system, meaning two commands per node.
Assuming you have your alignment commands in a file called aln.cmds, here's how to create and submit a batch job for the commands.
launcher_creator.py -n aln -j aln.cmds -t 12:00:00 -q normal -w 2 sbatch aln.slurm showq -u
Note the maximum run time specified here is 12 hours (-t 12:00:00). This is a reasonable value for a higher eukaryote with 20-40 M reads, and is way more than a yeast alignment would need (~ 4 hours). For very deeply sequenced eukaryotes (e.g. human genome re-sequencing with hundresd of millions of reads), you may want to specify the maximum job time of 48 hours.
Exercise: What would alignment commands look like if you were putting it in a batch system .cmds file?
x
Exercise #2: Bowtie2 global alignment - Vibrio cholerae RNA-seq
While we have focused on aligning eukaryotic data, the same tools can be used to perform identical functions with prokaryotic data. The major differences are less about the underlying data and much more about the external/public databases established to store and distribute reference data. For example, the Illumina iGenome resource provides pre-processed and uniform reference data, designed to be out-of-the-box compatible with aligners like bowtie2 and bwa. However, the limited number of available species are heavily biased towards model eukaryotes. If we wanted to study a prokaryote, the reference data must be downloaded from a resource like GenBank, and processed/indexed similarly to the procedure for mirbase.
While the alignment procedure for prokaryotes is broadly analogous, the reference preparation process is somewhat different, and will involve use of a biologically-oriented scripting library called BioPerl. In this exercise, we will use some RNA-seq data from Vibrio cholerae, published last year on GEO here, and align it to a reference genome.
Overview of Vibrio cholerae alignment workflow with Bowtie2
Alignment of this prokaryotic data follows the workflow below. Here we will concentrate on steps 1 and 2.
- Prepare the vibCho reference index for bowtie2 from a GenBank record using BioPerl
- Align reads using bowtie2, producing a SAM file
- Convert the SAM file to a BAM file (samtools view)
- Sort the BAM file by genomic location (samtools sort)
- Index the BAM file (samtools index)
- Gather simple alignment statistics (samtools flagstat and samtools idxstat)
Obtaining the GenBank record(s)
V. cholerae has two chromosomes. We download each separately.
- Navigate to http://www.ncbi.nlm.nih.gov/nuccore/NC_012582
- click on the Send down arrow (top right of page)
- select Complete Record
- select Clipboard as Destination
- click Add to Clipboard
- Perform these steps in your Terminal window
- mkdir $WORK/tmp; cd $WORK/tmp
- type wget then paste the URL from the clipboard
- Repeat steps 1 and 2 fot the 2nd chromosome
- NCBI URL is http://www.ncbi.nlm.nih.gov/nuccore/NC_012583
- you should now have 2 files, NC_012582 and NC_012583
- Combine the 2 files into one using cat
- cat NC_012582 NC_012583 > vibCho.gbk
Converting GenBank records into sequence (FASTA) and annotation (GFF) files
As noted earlier, many microbial genomes are available through repositories like GenBank that use specific file format conventions for storage and distribution of genome sequence and annotations. The GenBank file format is a text file that can be parsed to yield other files that are compatible with the pipelines we have been implementing.
Go ahead and look at some of the contents of a GenBank file with the following commands (execute these one at a time):
cd $WORK/core_ngs/references less vibCho.O395.gbk # use q to quit less grep -A 5 ORIGIN vibCho.O395.gbk
As the less command shows, the file begins with a description of the organism and some source information, and the contains annotations for each bacterial gene. The grep command shows that, indeed, there is sequence information here (flagged by the word ORIGIN) that could be exported into a FASTA file. There are a couple ways of extracting the information we want, namely the reference genome and the gene annotation information, but a convenient one (that is available through the module system at TACC) is BioPerl.
We load BioPerl like we have loaded other modules, with the caveat that we must load regular Perl before loading BioPerl:
module load perl module load bioperl
These commands make several scripts directly available to you. The one we will use is called bp_seqconvert.pl, and it is a BioPerl script used to inter-convert file formats like FASTA, GBK, and others. This script produces two output files:
- a FASTA format file for indexing and alignment
- a GFF file (standing for General Feature Format) contains information about all genes (or, more generally, features) in the genome
- remember, annotations such as GFFs must always match the reference you are using
To see how to use the script, just execute:
bp_seqconvert.pl
Clearly, there are many file formats that we can use this script to convert. In our case, we are moving from genbank to fasta, so the commands we would execute to produce and view the FASTA files would look like this:
cd $WORK/core_ngs/references bp_seqconvert.pl --from genbank --to fasta < vibCho.O395.gbk > vibCho.O395.fa mv vibCho.O395.fa fasta/ grep ">" fasta/vibCho.O395.fa less fasta/vibCho.O395.fa
Now we have a reference sequence file that we can use with the bowtie2 reference builder, and ultimately align sequence data against.
Recall from when we viewed the GenBank file that there are genome annotations available as well that we would like to extract into GFF format. However, the bp_seqconvert.pl script is designed to be used to convert sequence formats, not annotation formats. Fortunately, there is another script called bp_genbank2gff3.pl that can take a GenBank file and produce a GFF3 (the most recent format convention for GFF files) file. To run it and see the output, run these commands:
bp_genbank2gff3.pl --format Genbank vibCho.O395.gbk mv vibCho.O395.gbk.gff vibCho.O395.gff less vibCho.O395.gff
After the header lines, each feature in the genome is represented by a line that gives chromosome, start, stop, strand, and other information. Features are things like "mRNA," "CDS," and "EXON." As you would expect in a prokaryotic genome it is frequently the case that the gene, mRNA, CDS, and exon annotations are identical, meaning they share coordinate information. You could parse these files further using commands like grep and awk to extract, say, all exons from the full file or to remove the header lines that begin with #.
Introducing bowtie2
Go ahead and load the bowtie2 module so we can examine some help pages and options. To do that, you must first load the perl module, and then the a specific version of bowtie2.
module load perl module load bowtie/2.2.0
Now that it's loaded, check out the options. There are a lot of them! In fact for the full range of options and their meaning, Google "Bowtie2 manual" and bring up that page. The Table of Contents is several pages long! Ouch!
This is the key to using bowtie2 - it allows you to control almost everything about its behavior, but that also makes it is much more challenging to use than bwa – and it's easier to screw things up too!
Building the bowtie2 vibCho index
Before the alignment, of course, we've got to build a mirbase index using bowtie2-build (go ahead and check out its options). Unlike for the aligner itself, we only need to worry about a few things here:
- reference_in file is just the FASTA file containing mirbase v20 sequences
- bt2_index_base is the prefix of where we want the files to go
To build the reference index for alignment, we actually only need the FASTA file, since annotations are often not necessary for alignment. (This is not always true - extensively spliced transcriptomes requires splice junction annotations to align RNA-seq data properly, but for now we will only use the FASTA file.)
mkdir -p $WORK/core_ngs/references/bt2/vibCho mv $WORK/core_ngs/references/vibCho.O395.fa $WORK/core_ngs/references/fasta cd $WORK/core_ngs/references/bt2/vibCho ln -s -f ../../fasta/vibCho.O395.fa ls -la
Now build the index using bowtie2-build:
bowtie2-build vibCho.O395.fa vibCho.O395
This should also go pretty fast. You can see the resulting files using ls like before.
Performing the bowtie2 alignment
Now we will go back to our scratch area to do the alignment, and set up symbolic links to the index in the work area to simplify the alignment command:
cd $SCRATCH/core_ngs/alignment ln -s -f $WORK/core_ngs/references/bt2/vibCho vibCho
Note that here the data is from standard mRNA sequencing, meaning that the DNA fragments are typically longer than the reads. There is likely to be very little contamination that would require using a local rather than global alignment, or many other pre-processing steps (e.g. adapter trimming). Thus, we will run bowtie2 with default parameters, omitting options other than the input, output, and reference index.
As you can tell from looking at the bowtie2 help message, the general syntax looks like this:
bowtie2 [options]* -x <bt2-idx> {-1 <m1> -2 <m2> | -U <r>} [-S <sam>]
So our command would look like this:
bowtie2 -x vibCho/vibCho.O395 -U fastq/cholera_rnaseq.fastq.gz -S cholera_rnaseq.sam
Create a commands file called bt2_vibCho.cmds with this task definition then generate and submit a batch job for it (time 1 hour, development queue).
When the job is complete you should have a cholera_rnaseq.sam file that you can examine using whatever commands you like.