Overview
In this lab, we'll look at how to identify enriched gene ontology (GO) terms. For this analysis, we'll be using the differential analysis results we generated using DESeq.
Introduction
WHAT ARE GO TERMS?
GO terms provide a standardized vocabulary to describe genes and gene products from different species. GO terms allow us to assign functionality to genes. The following properties are described for gene products:
- cellular component, describes where in a cell a gene acts, what cellular unit the gene is part of
- molecular function, describes the function carried out by the gene, such as binding or catalysis;
- biological process, a set of molecular functions, with a defined beginning and end, makes up a biological process. This describes biological phenomenon like DNA replication.
All GO terms have an ID that looks like GO:0006260 and a name like DNA replication.
All GO terms have alist of genes that belong to that particular term.
GO terms are hierarchical consisting of broader parent GO terms and narrower child GO terms. For example, DNA replication is a child of GO term:cellular metabolic process. DNA replication has child GO terms like regulation of DNA replication, strand elongation.
WHAT IS GO ENRICHMENT?
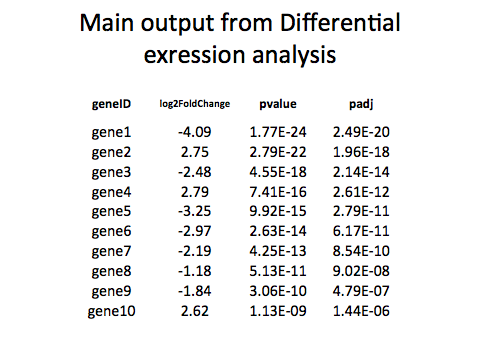
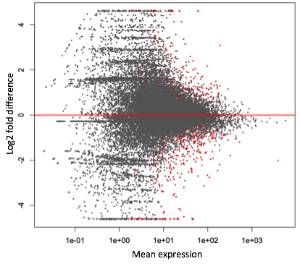
GO enrichment is a way of summarizing the FUNCTIONS AND TYPES of genes that are differentially expressed.
CLASSICAL GO ENRICHMENT
Input:
- A. Total number of genes we are looking at (ALL genes).
- B. Number of genes of interest, that is, in our DEG list (DEG).
- C. Total number of genes in the GO term
- D. Number of genes from our genes of interest (DEG) that are also in the GO term.
Enrichment test: whether “DEG list” contain more representatives of a certain GO category than expected by chance (Fisher’s exact, hypergeometric, or similar test).
If the number of genes from our list that belong to GO term GO:0001 (D) is significant compared to the total number of genes in that GO term (C) and the total number of genes in our experiment (A), we consider that GO term to be enriched in our data.
RANK-BASED GO ENRICHMENT
Input:To avoid enforcing arbitrary cutoffs, input all genes, ranked by something (like foldchange, pvalue).
Enrichment test: whether a GO category is significantly enriched within top ranking genes.
WHAT DOES THIS MEAN TO US?
Many genes may be changing, but they may all be linked to similar biological processes. From a list of changing genes -> list of affected biologial processes. We can better elucidate the biological events that are represented by our differential gene finding.
We also reduce the dataset considerably- from large number of genes to a smaller number of functions/processes.
We go from up and downregulated genes between two conditions to up and down regulated processes between two conditions.
TOOLS AVAILABLE FOR GO/PATHWAY ENRICHMENT
R package: GOSeq, topGO
Web-based tool for GO enrichment: Gorilla
Web-based tool for GO/pathway enrichment: Enrichr
Run Gorilla- Classical enrichment
#Make sure you are in the right location cds cd my_rnaseq_course/day_4_partA/go_enrichment
GET ALL INPUT:
Get all input files from DESeq2 output:
"","id","baseMean","baseMeanA","baseMeanB","foldChange","log2FoldChange","pval","padj"
"131","FBgn0000370",7637.91654540105,4217.77033402576,11058.0627567763,2.62177925326286,1.39054621964443,1.2887282997047e-116,7.22484613473489e-113
"2489","FBgn0025682",6038.35042952997,3300.21617337019,8776.48468568974,2.65936660649935,1.41108267336748,1.36704751839828e-116,7.22484613473489e-113
......
INPUT FILE 1: DEG (contains the 76 genes that meet our fold change an p value cut offs)
FBgn0000370
FBgn0025682
FBgn0086904
#Alter this old command to pull out Gene ids corresponding to DEGs and store it in a file called DEG
sed 's/,/\t/g' deseq2_htseq_C1_vs_C2.csv|awk '{if ((($3>=1)||($3<=-1))&&($6<=0.05)) print $1}' |sed 's/"//g'|grep '^FB' > DEG
.....
INPUT FILE 2: ALL (contains all 14869 genes)
FBgn0000370
FBgn0025682
FBgn0086904
.....
#Command to pull out ALL gene ids and store it in a file called ALL sed 's/,/\t/g' deseq2_htseq_C1_vs_C2.csv|cut -f 1|sed 's/"//g'|grep '^FB' > ALL
SCP THE DATA OVER TO YOUR COMPUTER:
#ON stampede2: copy the path for the ALL and DEG files pwd #ON LOCAL COMPUTER: from a terminal tab scp <username>@stampede2.tacc.utexas.edu:<pathtofileson/DEG> . scp <username>@stampede2.tacc.utexas.edu:<pathtofileson/ALL> .
RUN GORILLA USING THE UNRANKED METHOD: http://cbl-gorilla.cs.technion.ac.il/
Run Gorilla- Rank based enrichment
INPUT FILE: ALLRANKED (all genes, ranked by adjusted pvalue)
FBgn0000370
FBgn0025682
FBgn0086904
...
##Command to pull out ALL gene ids, sorted by adjpvalue store it in a file called ALLRANKED #Remember we already sorted our results by adjusted pvalue in the deseq2 script before writing it out to a file. So #you just need to pull out the gene ids in the order it already is in. sed 's/,/\t/g' deseq2_htseq_C1_vs_C2.csv|cut -f 1|sed 's/"//g'|grep '^FB' > ALLRANKED
SCP THE DATA OVER TO YOUR COMPUTER:
#ON stampede2: copy the path for the ALLRANKED file pwd #ON LOCAL COMPUTER: from a terminal tab scp <username>@stampede2.tacc.utexas.edu:<pathtofileson/ALLRANKED> .
RUN GORILLA USING THE RANKED METHOD: http://cbl-gorilla.cs.technion.ac.il/
Go back to COURSE OUTLINE