| Tip | ||
|---|---|---|
| ||
Our TACC allocation/project for this class is UT-2015-05-18. Use our summer school reservation (CoreNGS-Tue) when submitting batch jobs to get higher priority on the Lonestar5 ls6 normal queue during this course today:
Note that the reservation name (CoreNGS-Tue) is different from the TACC allocation/project for this class, which is OTH21164. |
| Table of Contents |
|---|
| Anchor | ||||
|---|---|---|---|---|
|
When you SSH into ls5 ls6, your session is assigned to one of a small set of login nodes (also called head nodes). These are not separate from the cluster compute nodes that will run your jobs.
Think of a node as a computer, like your laptop, but probably with more cores and memory. Now multiply that computer a thousand or more, and you have a cluster.
...
The small set of login nodes are a shared resource (type the users command to see everyone currently logged in) and are not meant for running interactive programs – for that you submit a description of what you want done to a batch system, which farms distributes the work out to one or more compute nodes.
On the other hand, the login nodes are intended for copying files to and from TACC, so they have a lot of network bandwidth while compute nodes have limited network bandwidth.
...
- Do not perform substantial computation on the login nodes.
- They are closely monitored, and you will get warnings from the TACC admin folks!
- Code is usually developed and tested somewhere other than TACC, and only moved over when pretty solid.
- Do not perform significant network access from your batch jobs.
- Instead, stage your data onto $SCRATCH from a login node onto $SCRATCH before submitting your job.
...
Lonestar6 and Stampede2 overview and comparison
Here is a comparison of the configurations and ls5 ls6 and stampede2. As you can see, stampede2 is the larger cluster, just recently launched in 2017., but ls6, launched om 2022, has fewer but more powerful nodes.
| ls6 | stampede2 | |||
|---|---|---|---|---|
| ls5 | stampede2 | login nodes | 63 20 128 cores each | 6 28 cores each |
| standard compute nodes | 560 AMD Epyc Milan processors 128 1,252 24 cores per node (48 virtual) | 4,200 KNL (Knights Landing) processors
1,736 SKX (Skylake) processors
| ||
| large memory GPU nodes | 10 total 2 w/1 TB memory, 48 cores | 16 AMD Epyc Milan processors 128 cores per nod 2x NVIDIA A100 GPUs | --Coming in 2018 | |
| batch system | SLURM | SLURM | ||
| maximum job run time | 48 hours, normal queue 2 hours, development queue | 96 hours on KNL nodes, normal queue 48 hours on SKX nodes |
Note the use of the term virtual core above. Compute cores are standalone processors – mini CPUs, each of which can execute separate sets of instructions. However modern cores may also have hyperthreading enabled, where a single core can appear as more than one virtual processor to the Operating system (see https://en.wikipedia.org/wiki/Hyper-threading for more on hyperthreading). For example, Lonestar5 nodes have 2 hyperthreads (HTs) per core, so they actually physically have only 24 cores per node, each of which has 2 HTs for a total of 48 virtual cores.
User guides for ls5 and stampede2 can be found at:
- https://portal.tacc.utexas.edu/user-guides/stampede2
- https://portal.tacc.utexas.edu/user-guides/lonestar5
, normal queue 2 hours, development queue |
User guides for ls6 and stampede2 can be found at:
- https://portal.tacc.utexas.edu/user-guides/lonestar6
- https://portal.tacc.utexas.edu/user-guides/stampede2
Unfortunately, the TACC user guides Unfortunately, the TACC user guides are aimed towards a different user community – the weather modelers and aerodynamic flow simulators who need very fast matrix manipulation and other high performance computing High Performance Computing (HPC) features. The usage patterns for bioinformatics – generally running 3rd party tools on many different datasets – datasets – is rather a special case for HPC.
Software at TACC
Programs and your $PATH
When you type in the name of an arbitrary progam (ls for example), how does the shell know where to find that program? The answer is your $PATH. $PATH is a pre-defined environment variable whose value is a list of directories.The shell looks for program names in that list, in the order the directories appear.
To determine where the shell will find a particular program, use the which command:
| Code Block | ||||
|---|---|---|---|---|
| ||||
which rsync
which cat |
The module system
The module system is an incredibly powerful way to have literally thousands of software packages available, some of which are incompatible with each other, without causing complete havoc. The TACC staff builds the desired package from source code in well-known locations that are NOT on your $PATH. Then, when a module is loaded, its binaries are added to your $PATH.
For example, the following module load command makes the bwa aligner available to you:
| Code Block | ||||
|---|---|---|---|---|
| ||||
# first type "bwa" to show that it is not present in your environment:
bwa
# it's not on your $PATH either:
which bwa
# now add bwa to your environment and try again:
module load bwa
bwa
# and see how it's now on your $PATH:
which bwa
# you can see the new directory at the front of $PATH
echo $PATH
# to remove it, use "unload"
module unload bwa
bwa
# gone from $PATH again...
which bwa |
module spider
These days the TACC module system includes hundreds of useful bioinformatics programs. To see if your favorite software package has been installed at TACC, use module spider:
| Code Block | ||
|---|---|---|
| ||
module spider samtools
module spider tophat
module spider bedtools
module spider GATK |
installing custom software
Even with all the tools available at TACC, inevitably you'll need something they don't have. In this case you can build the tool yourself and install it in a local TACC directory. While building 3rd party tools is beyond the scope of this course, it's really not that hard. The trick is keeping it all organized.
For one thing, remember that your $HOME directory quota is fairly small (10 GB on ls5), and that can fill up quickly if you install many programs. We recommend creating an installation area in your $WORK directory and installing programs there. You can then make symbolic links to the binaries you need in your $HOME/local/bin directory (which was added to your $PATH in your .bashrc).
See how we used a similar trick to make the launcher_maker.py program available to you. Using the ls -l option shows you where symbolic links point to:
| Code Block | ||||
|---|---|---|---|---|
| ||||
ls -l ~/local/bin |
| Expand | ||
|---|---|---|
| ||
|
| Warning | ||
|---|---|---|
| ||
Remember that the order of locations in the $PATH environment variable is the order in which the locations will be searched. In particular, the module load command adds to the front of your path. This can mask similarly-named programs, for example, in your $HOME/local/bin directory. |
...
Job execution is controlled by the SLURM batch system on both ls5 and stampede2.
To run a job you prepare 2 files:
- a commands file file containing the commands to run, one command per line (<job_name>.cmds)
- a job control file that describes how to run the job (<job_name>.slurm)
The process of running the job involves these steps:
- Create a commands file containing exactly one command per line.
- Prepare a job control file for the commands file that describes how the job should be run.
- You submit the job control file to the batch system. The job is then said to be queued to run.
- The batch system prioritizes the job based on the number of compute nodes needed and the job run time requested.
- When compute nodes become available, the job tasks (command lines in the <job_name>.cmds file) are assigned to one or more compute nodes and begin to run in parallel.
- The job completes when either:
- you cancel the job manually
- all tasks in the job complete (successfully or not!)
- the requested job run time has expired
SLURM at a glance
Here are the main components of the SLURM batch system.
...
TACC calls our type of processing "parameter sweep jobs" and has a special process for running them, using their launcher module.
About cores and hyperthreads
Note the use of the term virtual core on stampede2. Compute cores are standalone processors – mini CPUs, each of which can execute separate sets of instructions. However modern cores may also have hyperthreading enabled, where a single core can appear as more than one virtual processor to the operating system (see https://en.wikipedia.org/wiki/Hyper-threading). For example, stampede2 nodes have 2 or 4 hyperthreads (HTs) per core. So KNL nodes with 4 HTs for each of the 68 physical cores, have a total of 272 virtual cores.
Threading is an operating system scheduling mechanism for allowing one CPU/core to execute multiple computations, seemingly in parallel.
The writer of a program that takes advantage of threading first identifies portions of code that can run in parallel because the computations are independent. The programmer assigns some number of threads to that work (usually based on a command-line option) using specific thread and synchronization programming language constructs. An example is the the samtools sort -@ N option to specify N threads can be used for sorting independent sets of the input alignments.
If there are multiple cores/CPUs available, the operating system can assign a program thread to each of them for actual parallelism. But only "seeming" (or virtual) parallelism occurs if there are fewer cores than the number of threads specified.
Suppose there's only one core/CPU. The OS assigns program thread A to the core to run until the program performs an I/O operation that causes it to be "suspended" for the I/O operation to complete. During this time, when normally the CPU would be doing nothing but waiting on the I/O to complete, the OS assigns program thread B to the CPU and lets it do some work. This threading allows more efficient use of existing cores as long as the multiple program threads being assigned do some amount of I/O or other operations that cause them to suspend. But trying to run multiple compute-only, no-I/O programs using multiple threads on one CPU just causes "thread thrashing" -- OS scheduler overhead when threads are suspended for time, not just I/O.
The analogy is a grocery store where there are 5 customers (threads). If there are 5 checkout lines (cores), each customer (thread) can be serviced in a separate checkout line (core). But if there's only one checkout line (core) open, the customers (threads) will have to wait in line. To be a more accurate analogy, any checkout clerk would be able to handle some part of checkout for each customer, then while waiting for the customer to find and enter credit card information, the clerk could handle a part of a different customer's checkout.
Hyperthreading is just a hardware implementation of OS scheduling. Each CPU offers some number of "virtual cores" (hyperthreads) that can "almost" act like separate cores using various hardware tricks. Still, if the work assigned to multiple hyperthreads on a single core does not pause from time to time, thread thrashing will occur.
Software at TACC
Programs and your $PATH
When you type in the name of an arbitrary program (ls for example), how does the shell know where to find that program? The answer is your $PATH. $PATH is a predefined environment variable whose value is a list of directories.The shell looks for program names in that list, in the order the directories appear.
To determine where the shell will find a particular program, use the which command. Note that which tells you where it looked if it cannot find the program.
| Code Block | ||||
|---|---|---|---|---|
| ||||
which rsync
which cat
which bwa # not yet available to you |
The module system
The module system is an incredibly powerful way to have literally thousands of software packages available, some of which are incompatible with each other, without causing complete havoc. The TACC staff stages packages in well-known locations that are NOT on your $PATH. Then, when a module is loaded, its binaries are added to your $PATH.
For example, the following module load command makes the singularity container management system available to you
Simple example
Let's go through a simple example. Execute the following commands to copy a pre-made simple.cmds commands file:
| Code Block | ||||
|---|---|---|---|---|
| ||||
mkdir -p $SCRATCH/core_ngs/slurm/simple
cd $SCRATCH/core_ngs/slurm/simple
cp $CORENGS/tacc/simple.cmds . |
What are the tasks we want to do? Each task corresponds to one line in the simple.cmds file, so let's take a look at it using the cat (concatenate) command that simply reads a file and writes each line of content to standard output (here, your Terminal):
| Code Block | ||||
|---|---|---|---|---|
| ||||
cat simple.cmds |
The tasks we want to perform look like this:
| Code Block | ||
|---|---|---|
| ||
echo "Command 1 on `hostname` - `date`" > cmd1.log 2>&1
echo "Command 2 on `hostname` - `date`" > cmd2.log 2>&1
echo "Command 3 on `hostname` - `date`" > cmd3.log 2>&1
echo "Command 4 on `hostname` - `date`" > cmd4.log 2>&1
echo "Command 5 on `hostname` - `date`" > cmd5.log 2>&1
echo "Command 6 on `hostname` - `date`" > cmd6.log 2>&1
echo "Command 7 on `hostname` - `date`" > cmd7.log 2>&1
echo "Command 8 on `hostname` - `date`" > cmd8.log 2>&1 |
There are 8 tasks. Each is a simple echo command that just outputs string containing the task number and date to a different file.
Use the handy launcher_maker.py program to create the job submission script.
| Code Block | ||||
|---|---|---|---|---|
| ||||
launcher_maker.py -n simple.cmds -t 0:05 -w 8 -v -a UT-2015-05-18 -q dev |
You should see output something like the following, and you should see a simple.slurm batch submission file in the current directory.
| Code Block |
|---|
launcher_maker.py (2016.02.21)
Job Parameters for ls5 on slurm:
Project: simple
Job file: simple.cmds
Batch file: simple.slurm (launcher v3)
Directory: /scratch/01063/abattenh/core_ngs/simple
Queue: development
Time: 00:05:00
No job notification email
Allocation: UT-2015-05-18
Job file has 8 command lines
Commands per node (wayness): 8
Total nodes: 1
Total cores: 24
Modules:
Depends on jobid: None
Type "sbatch simple.slurm" to queue your job |
Submit your batch job like this, then check the batch queue to see the job's status.
| Code Block | ||||
|---|---|---|---|---|
| ||||
sbatch simple.slurm
showq -u |
If you're quick, you'll see a queue status something like this:
| Code Block |
|---|
SUMMARY OF JOBS FOR USER: <abattenh>
ACTIVE JOBS--------------------
JOBID JOBNAME USERNAME STATE CORE REMAINING STARTTIME
================================================================================
1578594 simple abattenh Running 48 0:04:52 Thu May 17 00:05:05
WAITING JOBS------------------------
JOBID JOBNAME USERNAME STATE CORE WCLIMIT QUEUETIME
================================================================================
Total Jobs: 1 Active Jobs: 1 Idle Jobs: 0 Blocked Jobs: 0 |
Notice in my queue status, where the STATE is Running, there are 48 COREs assigned. Why is this, since there were only 8 tasks?
The answer is that the batch jobs cannot share a node – every job, no matter how few tasks requested, will be assigned at least one node. And ls5 nodes have 48 virtual cores each. So the number of cores used will always be an even multiple of 48.
If you don't see your simple job in either the ACTIVE or WAITING sections of your queue, it probably already finished – it should only run for a second or two!
Exercise: What files were created by your job?
| Expand | ||
|---|---|---|
| ||
ls should show you something like this:
The newly created files are the .log files, as well as simple.1578594.joblog. |
filename wildcarding
Here's a cute trick for viewing the contents all your output files at once, using the cat command and filename wildcarding.
| Code Block | ||||
|---|---|---|---|---|
| ||||
cat cmd*.log |
The cat command actually takes a list of one or more files (if you're giving it files rather than standard input – more on this shortly) and outputs the concatenation of them to standard output. The asterisk ( * ) in cmd*.log is a multi-character wildcard that matches any filename starting with cmd then ending with .log. So it would match cmd_hello_world.log. You can also specify single-character matches inside brackets ( [ ] ) in either of these ways, this time using the ls command so you can better see what is matching:
| Code Block | ||||
|---|---|---|---|---|
| ||||
ls cmd[12345678].log
ls cmd[1-8].log |
This technique is sometimes called filename globbing, and the pattern a glob. Don't ask me why – it's a Unix thing. Globbing – translating a glob pattern into a list of files – is one of the handy thing the bash shell does for you. (Read more about Wildcards and special filenames)
Exercise: How would you list all files starting with simple?
| Expand | ||
|---|---|---|
| ||
ls simple* |
Here's what my cat output looks like. Notice the times are all the same, because all the tasks ran in parallel. That's the power of cluster computing!
| Code Block |
|---|
Command 1 on nid00008 - Thu May 17 00:05:13 CDT 2018
Command 2 on nid00008 - Thu May 17 00:05:13 CDT 2018
Command 3 on nid00008 - Thu May 17 00:05:13 CDT 2018
Command 4 on nid00008 - Thu May 17 00:05:13 CDT 2018
Command 5 on nid00008 - Thu May 17 00:05:13 CDT 2018
Command 6 on nid00008 - Thu May 17 00:05:13 CDT 2018
Command 7 on nid00008 - Thu May 17 00:05:13 CDT 2018
Command 8 on nid00008 - Thu May 17 00:05:13 CDT 2018 |
echo
| |
# first type "singularity" to show that it is not present in your environment:
singularity
# it's not on your $PATH either:
which singularity
# now add biocontainers to your environment and try again:
module load biocontainers
# and see how singularity is now on your $PATH:
which singularity
# you can see the new directory at the front of $PATH
echo $PATH
# to remove it, use "unload"
module unload biocontainers
singularity
# gone from $PATH again...
which singularity |
TACC BioContainers modules
It is quite a large systems administration task to install software at TACC and configure it for the module system. As a result, TACC was always behind in making important bioinformatics software available. To address this problem, TACC moved to providing bioinformatics software via containers, which are virtual machines like VMware and Virtual Box, but are lighter weight: they require less disk space because they rely more on the host's base Linux environment. Specifically, TACC (and many other High Performance Computing clusters) use Singularity containers, which are similar to Docker containers but are more suited to the HPC environment (in fact one can build a Docker container then easily convert it to Singularity for use at TACC).
TACC obtains its containers from BioContainers (https://biocontainers.pro/ and https://github.com/BioContainers/containers), a large public repository of bioinformatics tool Singularity containers. This has allowed TACC to easily provision thousands of such tools!
These BioContainers are not visible in TACC's "standard" module system, but only after the master biocontainers module is loaded. Once it has been loaded, you can search for your favorite bioinformatics program using module spider.
| Code Block | ||
|---|---|---|
| ||
# Verify that samtools is not available
samtools
# and cannot be found in the standard module system
module spider samtools
# Load the BioContainers master module (this takes a while)
module load biocontainers
# Now look for these programs
module spider samtools
module spider Rstats
module spider kallisto
module spider bowtie2
module spider minimap2
module spider multiqc
module spider gatk
module spider velvet |
Notice how the BioContainers module names have "ctr" in their names, version numbers, and other identifying information.
| Tip |
|---|
The standard TACC module system has been phased out for bioinformatics programs, so always look for your application in BioContainers. While it's great that there are now hundreds of programs available through BioContainers, the one drawback is that they can only be run on cluster compute nodes, not on login nodes. To test BioContainer program interactively, you will need to use TACC's idev command to obtain an interactive cluster node. More on this shortly... |
loading a biocontainer module
Once the biocontainers module has been loaded, you can just module load the desired tool, as with the kallisto pseudo-aligner program below.
| Code Block | ||
|---|---|---|
| ||
# Load the Biocontainers master module
module load biocontainers
# Verify kallisto is not yet available
kallisto
# Load the default kallisto biocontainer
module load kallisto
# Verify kallisto is not available (although not on login nodes)
kallisto |
Note that loading a BioContainer does not add anything to your $PATH. Instead, it defines an alias, which is just a shortcut for executing the command. You can see the alias definition using the type command. And you can ensure the program is available using the command -v utility.
| Code Block | ||
|---|---|---|
| ||
# Note that kallisto has not been added to your $PATH, but instead has an alias
which kallisto
# Ensure kallisto is available with command -v
command -v kallisto |
installing custom software
Even with all the tools available at TACC, inevitably you'll need something they don't have. In this case you can build the tool yourself and install it in a local TACC directory. While building 3rd party tools is beyond the scope of this course, it's really not that hard. The trick is keeping it all organized.
For one thing, remember that your $HOME directory quota is fairly small (10 GB on ls6), and that can fill up quickly if you install many programs. We recommend creating an installation area in your $WORK directory and installing programs there. You can then make symbolic links to the binaries you need in your ~/local/bin directory (which was added to your $PATH in your .bashrc).
See how we used a similar trick to make the launcher_creator.py program available to you. Using the ls -l option shows you where symbolic links point to:
| Code Block | ||||
|---|---|---|---|---|
| ||||
ls -l ~/local/bin
# this will tell you the real location of the launcher_creator.py script is
# /work/projects/BioITeam/common/bin/launcher_creator.py |
| Warning | ||
|---|---|---|
| ||
Remember that the order of locations in the $PATH environment variable is the order in which the locations will be searched. |
Job Execution
Job execution is controlled by the SLURM batch system on both stampede2 and ls6.
To run a job you prepare 2 files:
- a commands file file containing the commands to run, one task per line (<job_name>.cmds)
- a job control file that describes how to run the job (<job_name>.slurm)
The process of running the job involves these steps:
- Create a commands file containing exactly one task per line.
- Prepare a job control file for the commands file that describes how the job should be run.
- You submit the job control file to the batch system. The job is then said to be queued to run.
- The batch system prioritizes the job based on the number of compute nodes needed and the job run time requested.
- When compute nodes become available, the job tasks (command lines in the <job_name>.cmds file) are assigned to one or more compute nodes and begin to run in parallel.
- The job completes when either:
- you cancel the job manually
- all job tasks complete (successfully or not!)
- the requested job run time has expired
SLURM at a glance
Here are the main components of the SLURM batch system.
| stampede2, ls5 | |
|---|---|
| batch system | SLURM |
| batch control file name | <job_name>.slurm |
| job submission command | sbatch <job_name>.slurm |
| job monitoring command | showq -u |
| job stop command | scancel -n <job name> |
Simple example
Let's go through a simple example. Execute the following commands to copy a pre-made simple.cmds commands file:
| Code Block | ||||
|---|---|---|---|---|
| ||||
mkdir -p $SCRATCH/core_ngs/slurm/simple
cd $SCRATCH/core_ngs/slurm/simple
cp $CORENGS/tacc/simple.cmds . |
What are the tasks we want to do? Each task corresponds to one line in the simple.cmds commands file, so let's take a look at it using the cat (concatenate) command that simply reads a file and writes each line of content to standard output (here, your Terminal):
| Code Block | ||||
|---|---|---|---|---|
| ||||
cat simple.cmds |
The tasks we want to perform look like this:
| Code Block | ||||
|---|---|---|---|---|
| ||||
sleep 5; echo "Command 1 on `hostname` - `date`" > cmd1.log 2>&1
sleep 5; echo "Command 2 on `hostname` - `date`" > cmd2.log 2>&1
sleep 5; echo "Command 3 on `hostname` - `date`" > cmd3.log 2>&1
sleep 5; echo "Command 4 on `hostname` - `date`" > cmd4.log 2>&1
sleep 5; echo "Command 5 on `hostname` - `date`" > cmd5.log 2>&1
sleep 5; echo "Command 6 on `hostname` - `date`" > cmd6.log 2>&1
sleep 5; echo "Command 7 on `hostname` - `date`" > cmd7.log 2>&1
sleep 5; echo "Command 8 on `hostname` - `date`" > cmd8.log 2>&1 |
There are 8 tasks. Each task sleeps for 5 seconds, then uses the echo command to output a string containing the task number and date to a log file named for the task number. Notice that we can put two commands on one line if they are separated by a semicolon ( ; ).
Use the handy launcher_creator.py program to create the job control Lets take a closer look at a typical task in the simple.cmds file.
| Code Block | ||||
|---|---|---|---|---|
| ||||
echo "Command 3 `date`" > cmd3.log 2>&1 |
The echo command is like a print statement in the bash shell. Echo takes its arguments and writes them to one line of standard output. While not always required, it is a good idea to put the output string in double quotes.
backtick evaluation
| |
launcher_creator.py -j simple.cmds -n simple -t 00:01:00 -a OTH21164 -q development |
You should see output something like the following, and you should see a simple.slurm batch submission file in the current directory.
| Code Block |
|---|
Project simple.
Using job file simple.cmds.
Using development queue.
For 00:01:00 time.
Using OTH21164 allocation.
Not sending start/stop email.
Launcher successfully created. Type "sbatch simple.slurm" to queue your job. |
Submit your batch job then check the batch queue to see the job's status.So what is this funny looking `date` bit doing? Well, date is just another Linux command (try just typing it in). Here we don't want the shell to put the string "date" in the output, we want it to execute the date command and put that result into the output. The backquotes ( ` ` also called backticks) around the date command tell the shell we want that command executed and its output substituted into the string. (Read more about Quoting in the shell.)
| Code Block | |||||
|---|---|---|---|---|---|
| |||||
sbatch simple.slurm showq -u# These are equivalent: date echo `date` # ButOutput looks differentsomething from this: echo date |
output redirection
There's still more to learn from one of our simple tasks, something called output redirection:
| Code Block | ||
|---|---|---|
| ||
echo "Command 3 `date`" > cmd3.log 2>&1 |
Normally echo writes its string to standard output. If you invoke echo in an interactive shell like Terminal, standard output is displayed to the Terminal window.

Usually we want to separate the outputs of all our commands. Why is this important? Suppose we run a job with 100 commands, each one a whole pipeline (alignment, for example). 88 finish fine but 12 do not. Just try figuring out which ones had the errors, and where the errors occurred, if all the output is in one intermingled file and all the error in another intermingled file!
So in the above example the first '>' says to redirect the standard output of the echo command to the cmd3.log file. The '2>&1' part says to redirect standard error to the same place. Technically, it says to redirect standard error (built-in Linux stream 2) to the same place as standard output (built-in Linux stream 1); and since standard output is going to cmd3.log, any standard error will go there also. (Read more about Standard I/O streams.)
So what happens when output is generated by tasks in a batch job? Well, you may have noticed the file winh a name like simple.1578594.joblog was created by your job. It contains all standard output and standard error, generated by your tasks that was not redirected elsewhere.
Job parameters
Now that we've executed a really simple job, let's take a look at some important job submission parameters. These correspond to arguments to the launcher_maker.py script.
A bit of background. Historically, TACC was set up to cater to researchers writing their own C or Fortran codes highly optimized to exploit parallelism (the HPC crowd). Much of TACC's documentation is aimed at this audience, which makes it difficult to pick out the important parts for us.
The kind of jobs we biologists generally run are relatively new to TACC. They even have special names for them: "parametric serial jobs" or "parametric sweeps", by which they mean the same program running on different data sets.
In fact there is a special software module required to run our jobs, called the launcher module. You don't need to worry about activating the launcher module; that's done by the <job_name>.slurm script created by launcher_maker.py like this:
| Code Block | ||
|---|---|---|
| ||
module load launcher |
The launcher module knows how to interpret various job parameters in the <job_name>.slurm batch SLURM submission script and use them to create your job and assign its tasks to compute nodes. Our launcher_maker.py program is a simple Python script that lets you specify job parameters and writes out a valid <job_name>.slurm submission script.
launcher_maker.py
If you call launcher_maker.py --help it gives you its usage description:
like this:
-------------------------------------------------------------
Welcome to the Lonestar6 Supercomputer
-------------------------------------------------------------
--> Verifying valid submit host (login1)...OK
--> Verifying valid jobname...OK
--> Verifying valid ssh keys...OK
--> Verifying access to desired queue (normal)...OK
--> Checking available allocation (OTH21164)...OK
Submitted batch job 232542
|
The queue status will show your job as ACTIVE while its running, or WAITING if not.
| Code Block |
|---|
SUMMARY OF JOBS FOR USER: <abattenh>
ACTIVE JOBS--------------------
JOBID JOBNAME USERNAME STATE NODES REMAINING STARTTIME
================================================================================
924965 simple abattenh Running 1 0:00:42 Sat Jun 3 21:33:31
WAITING JOBS------------------------
JOBID JOBNAME USERNAME STATE NODES WCLIMIT QUEUETIME
================================================================================
Total Jobs: 1 Active Jobs: 1 Idle Jobs: 0 Blocked Jobs: 0 |
If you don't see your simple job in either the ACTIVE or WAITING sections of your queue, it probably already finished – it should only run for a few seconds!
Notice in my queue status, where the STATE is Running, there is only one node assigned. Why is this, since there were 8 tasks?
Every job, no matter how few tasks requested, will be assigned at least one node. Each lonestar6 node has 128 physical cores, so each of the 8 tasks can be assigned to a different core.
Exercise: What files were created by your job?
| Expand | ||
|---|---|---|
| ||
ls should show you something like this:
The newly created files are the .log files, as well as error and output logs simple.e924965 and simple.o924965. |
filename wildcarding
You can look at one of the output log files like this:
| Code Block | ||
|---|---|---|
| ||
cat cmd1.log |
But here's a cute trick for viewing the contents all your output files at once, using the cat command and filename wildcarding.
| Code Block | ||||
|---|---|---|---|---|
| ||||
cat cmd*.log |
The cat command can take a list of one or more files. The asterisk ( * ) in cmd*.log is a multi-character wildcard that matches any filename starting with cmd then ending with .log.
You can also specify single-character matches inside brackets ( [ ] ) in either of the ways below, this time using the ls command so you can better see what is matching:
| Code Block | ||||
|---|---|---|---|---|
| ||||
ls cmd[1234].log
ls cmd[2-6].log |
This technique is sometimes called filename globbing, and the pattern a glob. Don't ask me why – it's a Unix thing. Globbing – translating a glob pattern into a list of files – is one of the handy thing the bash shell does for you. (Read more about Pathname wildcards)
Exercise: How would you list all files starting with "simple"?
| Expand | ||
|---|---|---|
| ||
ls simple* |
Here's what my cat output looks like. Notice the times are all nearly the same because all the tasks ran in parallel. That's the power of cluster computing!
| Code Block |
|---|
Command 1 on c304-005.ls6.tacc.utexas.edu - Sat Jun 3 21:33:50 CDT 2023
Command 2 on c304-005.ls6.tacc.utexas.edu - Sat Jun 3 21:33:44 CDT 2023
Command 3 on c304-005.ls6.tacc.utexas.edu - Sat Jun 3 21:33:46 CDT 2023
Command 4 on c304-005.ls6.tacc.utexas.edu - Sat Jun 3 21:33:47 CDT 2023
Command 5 on c304-005.ls6.tacc.utexas.edu - Sat Jun 3 21:33:51 CDT 2023
Command 6 on c304-005.ls6.tacc.utexas.edu - Sat Jun 3 21:33:47 CDT 2023
Command 7 on c304-005.ls6.tacc.utexas.edu - Sat Jun 3 21:33:51 CDT 2023
Command 8 on c304-005.ls6.tacc.utexas.edu - Sat Jun 3 21:33:49 CDT 2023 |
echo
Lets take a closer look at a typical task in the simple.cmds file.
| Code Block | ||||
|---|---|---|---|---|
| ||||
sleep 5; echo "Command 3 `date`" > cmd3.log 2>&1 |
The echo command is like a print statement in the bash shell. echo takes its arguments and writes them to standard output. While not always required, it is a good idea to put echo's output string in double quotes.
backtick evaluation
So what is this funny looking `date` bit doing? Well, date is just another Linux command (try just typing it in) that just displays the current date and time. Here we don't want the shell to put the string "date" in the output, we want it to execute the date command and put the result text into the output. The backquotes ( ` ` also called backticks) around the date command tell the shell we want that command executed and its standard output substituted into the string. (Read more about Quoting in the shell.)
| Code Block | ||||
|---|---|---|---|---|
| ||||
# These are equivalent:
date
echo `date`
# But different from this:
echo date |
output redirection
There's still more to learn from one of our simple tasks, something called output redirection.
Every command and Unix program has three "built-in" streams: standard input, standard output and standard error, each with a name, a number, and a redirection syntax.
Normally echo writes its string to standard output, but it could encounter an error and write an error message to standard error. We want both standard output and standard error for each task stored in a log file named for the command number.
| Code Block | ||||
|---|---|---|---|---|
| ||||
sleep 5; echo "Command 3 `date`" > cmd3.log 2>&1 |
So in the above example the first '>' says to redirect the standard output of the echo command to the cmd3.log file. The '2>&1' part says to redirect standard error to the same place. Technically, it says to redirect standard error (built-in Linux stream 2) to the same place as standard output (built-in Linux stream 1); and since standard output is going to cmd3.log, any standard error will go there also. (Read more about Standard streams and redirection)
When the TACC batch system runs a job, all outputs generated by tasks in the batch job are directed to one output and error file per job. Here they have names like simple.e924965 and simple.o924965. simple.o924965 contains all standard output and simple.o924965 contains all standard error generated by your tasks that was not redirected elsewhere, as well as information relating to running your job and its tasks. For large jobs with complex tasks, it is not easy to troubleshoot execution problems using these files.
So a best practice is to separate the outputs of all our tasks into individual log files, one per task, as we do here. Why is this important? Suppose we run a job with 100 commands, each one a whole pipeline (alignment, for example). 88 finish fine but 12 do not. Just try figuring out which ones had the errors, and where the errors occurred, if all the standard output is in one intermingled file and all standard error in the other intermingled file!
Job parameters
Now that we've executed a really simple job, let's take a look at some important job submission parameters. These correspond to arguments to the launcher_creator.py script.
A bit of background. Historically, TACC was set up to cater to researchers writing their own C or Fortran codes highly optimized to exploit parallelism (the HPC crowd). Much of TACC's documentation is aimed at this audience, which makes it difficult to pick out the important parts for us.
The kind of jobs we biologists generally run are relatively new to TACC. They even have a special name for them: "parametric sweeps", by which they mean the same program running on different data sets.
In fact there is a special software module required to run our jobs, called the launcher module. You don't need to worry about activating the launcher module – that's done by the <job_name>.slurm script created by launcher_creator.py like this:
| Code Block | ||
|---|---|---|
| ||
module load launcher |
The launcher module knows how to interpret various job parameters in the <job_name>.slurm batch SLURM submission script and use them to create your job and assign its tasks to compute nodes. Our launcher_creator.py program is a simple Python script that lets you specify job parameters and writes out a valid <job_name>.slurm submission script.
launcher_creator.py
If you call launcher_creator.py with no arguments it gives you its usage description. Because it is a long help message, we may want to pipe the output to more, a pager that displays one screen of text at a time. Type the spacebar to advance to the next page, and Ctrl-c to exit from more.
| Code Block | ||||
|---|---|---|---|---|
| ||||
# Use spacebar to page forward; Ctrl-c to exit
launcher_creator.py | more |
| Code Block | ||
|---|---|---|
| ||
usage: launcher_creator.py [-h] -n NAME -t TIME_REQUEST [-j JOB_FILE]
[-b SHELL_COMMANDS] [-B SHELL_COMMANDS_FILE]
[-q QUEUE] [-a [ALLOCATION]] [-m MODULES]
[-M MODULES_FILE] [-w WAYNESS] [-N NUM_NODES]
[-e [EMAIL]] [-l LAUNCHER] [-s]
Create launchers for TACC clusters. Report problems to rt-
other@ccbb.utexas.edu
optional arguments:
-h, --help show this help message and exit
Required:
-n NAME, --name NAME The name of your job.
-t TIME_REQUEST, --time TIME_REQUEST
The time you want to give to your job. Format:
hh:mm:ss
Commands:
You must use at least one of these options to submit your commands for TACC.
-j JOB_FILE, --jobs JOB_FILE
The name of the job file containing your commands.
-b SHELL_COMMANDS, --bash SHELL_COMMANDS
A string of shell (Bash, zsh, etc) commands that are
executed before any parametric jobs are launched.
-B SHELL_COMMANDS_FILE, --bash_file SHELL_COMMANDS_FILE
A file containing shell (Bash, zsh, etc) commands that | ||
| Code Block | ||
usage: launcher_maker.py [-h] [-n NAME] [-t TIME] [-w {1,2,3,4,6,8,12,24}] [are executed before any parametric jobs are launched. Optional: -q {normal,dev,largemem,serial}] [-j JOB]QUEUE, --queue QUEUE The [-l LAUNCHER] [-m MODULES] [-b BASH_COMMANDS]TACC allocation for job submission. [-e EMAIL] [-d DEPENDS] [-a ALLOCATION] [-v] [-V] optional arguments: -hDefault="development" -a [ALLOCATION], -A [ALLOCATION], --helpallocation [ALLOCATION] show this help message and exit -n NAME The TACC allocation for job submission. You can Job name (required). Usually the name of your commands set a default ALLOCATION environment variable. file (with or without .cmds suffix) -t TIME-m MODULES, --modules MODULES Maximum job runA time. Default="02:00:00" (2 hrs). list of module commands. The "launcher" module is Format: [h]h (-t 6 =6always hrs)automatically or [h]h:mm (-t 12:30 =12 hrincluded. Example: -m "module 30swap min)intel or [h]h:mm:ss (-t 0:30 =30 min)gcc; module load bedtools" -w {1,2,3,4,6,8,12,24}M MODULES_FILE, --modules_file MODULES_FILE Wayness:A thefile numbercontaining ofmodule commands to give each node. -w WAYNESS, --wayness WAYNESS Default=24. -q {normal,dev,largemem,serial} Wayness: the number of commands you want to give each TACC queue for job submission. Default=normal. -j JOB node. The default is the number of cores per node. -N Name of the job file containing commands. NUM_NODES, --num_nodes NUM_NODES Number of nodes to Default="<name>.cmds" -l LAUNCHER request. You probably don't need Name of the launcher script to be created. this option. Use wayness instead. You ONLY need it if Default="<name>.slurm" -m MODULES Comma-separated list of moduleyou nameswant to load. Example: run a job list that isn't defined at the "bwa,samtools". -b BASH_COMMANDS time you Stringsubmit ofthe Bashlauncher. commands to execute before the job-e [EMAIL], --email [EMAIL] starts (enclose in single quotes). -e EMAIL Your email address if you want to receive an email Email address for job start/end notification. Can also from Lonestar when your job starts and ends. Without be specified in SEND_EMAIL environment variable. -a ALLOCATION an argument, it will use TACCa allocation for job submission. Use this ONLY if default EMAIL_ADDRESS environment variable. -l LAUNCHER, you have multiple projects. Can be specified in --launcher_name LAUNCHER The name of the launcher script that ALLOCATIONwill environmentbe variablecreated. -v, --verbose If present, echoes key submission info to stdout.Default="<name>.slurm" -V,s --version If present, prints program version then exits. |
Because it is a long help message, we may want to pipe the output to more, a pager that displays one screen of text at a time. Type the spacebar to advance to the next page, and Ctrl-c to exit from more.
| Code Block | ||||
|---|---|---|---|---|
| ||||
# Use spacebar to page forward; Ctrl-c to exit launcher_maker.py -h | more Echoes the launcher filename to stdout. |
| Expand | ||
|---|---|---|
| ||
| ||
| Expand | ||
| ||
launcher_maker.py and the older launcher_creator.py are both BioITeam programs that create batch submission scripts. They are quite similar in may ways, but also reflect the preferences of their two authors (Anna & Benni). We're using launcher_maker.py in this class because Anna developed it, and because she and Benni often disagree about programming approaches. This is not an uncommon phenomenon in the world of software development |
job name and commands file
...
| Code Block | ||||
|---|---|---|---|---|
| ||||
launcher_makercreator.py -nj simple.cmds -tn 0:05simple -w 8t 00:01:00 -v -a UT-2015-05-18OTH21164 -q devdevelopment |
- The name of your commands file is given with the -n <job_name>.cmdsj simple.cmds option.
- Your desired job name is given with the -n simple option argument.
- The <job_name> prefix (here simple) is the job name you will see in your queue.
- By default a corresponding <job_name>.slurm batch file is created for you.
- It contains the name of the commands file that the batch system will execute.
...
TACC resources are partitioned into queues: a named set of compute nodes with different characteristics. The major main ones on ls5 ls6 are listed below. Generally you use development (-q devdevelopment) when you are writing and testing your code, then normal once you're sure your commands will execute properly.
| queue name | maximum runtime | purpose | |||
|---|---|---|---|---|---|
| development | 2 hrs | development/testing and short jobs (typically has short queue wait times) | |||
| normal | 48 hrs | normal priority jobs (queue waits can sometimes be are often long) | largemem | 48 hrs | large memory jobs |
- In launcher_makercreator.py, the queue is specified by the -q argument.
- The default queue is normal development. Specify -q dev normal for development normal queue jobs.
- The maximum runtime you are requesting for your job is specified by the -t argument.
- Format is hh:mm:ss
- Note that your job will be terminated without warning at the end of its time limit!
...
You may be a member of a number of different projects, hence have a choice which resource allocation to run your job under.
- You specify that allocation name with the -a argument of launcher_makercreator.py.
- If you have set an $ALLOCATION environment variable to an allocation name, it that allocation will be used if you are a member of only one TACC Project.
| Expand | |||||||
|---|---|---|---|---|---|---|---|
| |||||||
The .bashrc login script you've installed for this course specifies the class's allocation as shown below. Note that this allocation will expire after the course, so you should change that setting appropriately at some point.
|
- When you run a batch job, your project allocation gets "charged" for the time your job runs, in the currency of SUs (System Units).
- SUs are related in some way to node hours, usually For most queues, 1 SU = 1 node / hour of compute time (large memory queues may charge more).
| Tip | ||
|---|---|---|
| ||
Jobs should consist of tasks that will run for approximately the same length of time. This is because the total node hours for your job is calculated as the run time for your longest running task (the one that finishes last). For example, if you specify 64 100 commands and 99 finish in 2 seconds but one runs for 24 hours, you'll be charged for 64 100 x 24 SUs ( node hours ) even though the total amount of work performed was only ~24 hours. |
...
One of the most confusing things in job submission is the parameter called wayness, which controls how many tasks are run on each computer compute node.
- Recall that there are 48 virtual 128 physical cores and 64 256 GB of memory on each compute node
- so technically theoretically you can could run up to 48 128 commands on a node, each with ~1.3 ~2 GB available memory
- you can usually run fewer tasks on a node, and if when you do, each task gets more resources
- Because bioinformatics programs generally require more memory and fewer cores, launcher_maker.py sets a 24 cores/node maximum.
| tasks per node (wayness) | cores available to each task | memory available to each task |
|---|---|---|
| 1 | 24128 | 64 ~256 GB |
| 2 | 1264 | 32 ~128 GB3 |
| 4 | 832 | 21.3~64 GB |
| 48 | 616 | 16 ~32 GB |
| 616 | 48 | 10.6 ~16 GB8 |
| 32 | 34 | 8~8 GB |
| 64 | 2 | 5.3~4 GB |
| 128 | 1 | 2.6~1 GB |
- In launcher_makercreator.py, wayness is specified by the -w argument.
- the default is 24 128 (one task per core)
- A special case is when you have only 1 command in your job.
- In that case, it doesn't matter what wayness you request.
- Your job will run on one compute node, and have all 48 cores available.
Your choice of the wayness parameter will depend on the nature of the work you are performing: its computational intensity, its memory requirements and its ability to take advantage of multi-processing/multi-threading (e.g. bwa -t option or tophat hisat2 -p option).
Wayness example
Let's use launcher_maker.py to explore wayness options. First copy over the wayness.cmds commands file:
| Code Block | ||||
|---|---|---|---|---|
| ||||
mkdir -p $SCRATCH/core_ngs/slurm/wayness
cd $SCRATCH/core_ngs/slurm/wayness
cp $CORENGS/tacc/wayness.cmds . |
| Tip |
|---|
Bioinformatics programs generally perform substantial I/O, require more memory and fewer cores, so you'll generally want to run only a few tasks per node. |
Wayness example
Let's use launcher_creator.py to explore wayness options. First copy over the wayness.cmds commands fileThe wayness.cmds commands file consists of 24 identical lines that look like this:
| Code Block | ||
|---|---|---|
| ||
sleep 3; echo "Command $LAUNCHER_JID of $LAUNCHER_NJOBS ($LAUNCHER_PPN per node) ran on node `hostname` core $LAUNCHER_TSK_ID." > cmd.$LAUNCHER_JID.log 2>&1 |
| Expand | ||
|---|---|---|
| ||
The wayness commands take advantage of a number of environment variables the launcher module system sets automatically for each task:
For more information, see https://github.com/TACC/launcher |
Create the batch submission script specifying a wayness of 8 (8 tasks per node), then submit the job and monitor the queue:
| Code Block | ||||
|---|---|---|---|---|
| ||||
launcher_maker.py -n wayness.cmds -w 8 -t 0:10 -v -a UT-2015-05-18 -q dev
sbatch wayness.slurm
showq -u |
Exercise: With 24 tasks requested and wayness of 8, how many nodes will this job require? How much memory will be allocated to each task?
| Expand | ||
|---|---|---|
| ||
3 nodes (24 tasks x 1 node/8 tasks) |
Exercise: If you specified a wayness of 2, how many nodes would this job require? How much memory could each task use?
| Expand | ||
|---|---|---|
| ||
12 nodes (24 tasks x 1 node/2 tasks) |
Look at the output file contents once the job is done.
| Code Block | ||
|---|---|---|
| ||
cat cmd*log
# or, for a listing ordered by command number (the 2nd field, a number)
cat cmd*log | sort -k 2,2n |
You should see something like output below.
| ||
# If $CORENGS is not defined:
export CORENGS=/work/projects/BioITeam/projects/courses/Core_NGS_Tools
cds
mkdir -p core_ngs/slurm/wayness
cd core_ngs/slurm/wayness
cp $CORENGS/tacc/wayness.cmds . |
Exercise: How many tasks are specified in the wayness.cmds file?
| Expand | ||
|---|---|---|
| ||
wc --help |
| Expand | |||||
|---|---|---|---|---|---|
| |||||
Find the number of lines in the wayness.cmds commands file using the wc (word count) command with the -l (lines) option:
The file has 16 lines, representing 16 tasks. |
The wayness.cmds commands file consists of a number of identical lines that look like this:
| Code Block | ||
|---|---|---|
| ||
sleep 3; echo "Command $LAUNCHER_JID of $LAUNCHER_NJOBS ($LAUNCHER_PPN per node) ran on node `hostname` core $LAUNCHER_TSK_ID" > cmd.$LAUNCHER_JID.log 2>&1 |
| Expand | ||
|---|---|---|
| ||
The wayness commands take advantage of a number of environment variables the launcher module system sets automatically for each task:
For more information, see https://github.com/TACC/launcher |
Create the batch submission script specifying a wayness of 4 (4 tasks per node), then submit the job and monitor the queue:
| Code Block | ||||
|---|---|---|---|---|
| ||||
launcher_creator.py -j wayness.cmds -n wayness -w 4 -t 00:02:00 -a OTH21164 -q development
sbatch wayness.slurm
showq -u |
Exercise: With 16 tasks requested and wayness of 4, how many nodes will this job require? How much memory will be available for each task?
| Expand | ||
|---|---|---|
| ||
4 nodes (16 tasks x 1 node/4 tasks) |
Exercise: If you specified a wayness of 2, how many nodes would this job require? How much memory could each task use?
| Expand | ||
|---|---|---|
| ||
8 nodes (16 tasks x 1 node/2 tasks) |
Look at the output file contents once the job is done.
| Code Block | ||
|---|---|---|
| ||
cat cmd*log
# or, for a listing ordered by node name (the 11th field)
cat cmd*log | sort -k 11,11 |
The vertical bar ( | ) above is the pipe operator, which connects one program's standard output to the next program's standard input.
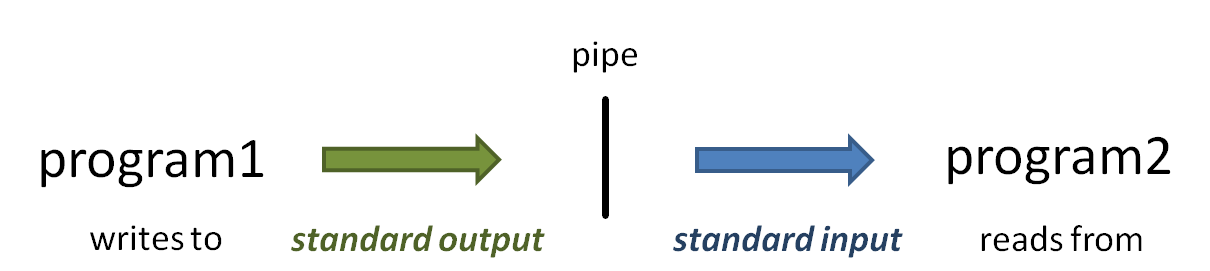
(Read more about the sort command at Linux fundamentals: cut, sort, uniq, and more about Piping)
You should see something like output below.
| Code Block | ||
|---|---|---|
| ||
Command 1 of 16 (4 | ||
| Code Block | ||
| ||
Command 1 of 24 (8 per node) ran on node nid00023 core 21. Command 2 of 24 (8 per node) ran on node nid00023 core 19. Command 3 of 24 (8 per node) ran on node nid00023 core 17. Command 4 of 24 (8 per node) ran on node nid00023 core 18. Command 5 of 24 (8 per node) ran on node nid00023 core 16. Command 6 of 24 (8 per node) ran on node nid00023 core 22. Command 7 of 24 (8 per node) ran on node nid00023 core 20. Command 8 of 24 (8 per node) ran on node nid00023c303-005.ls6.tacc.utexas.edu core 23.0 Command 910 of 2416 (84 per node) ran on node nid00022c304-005.ls6.tacc.utexas.edu core 14.9 Command 1011 of 2416 (84 per node) ran on node nid00022c304-005.ls6.tacc.utexas.edu core 10. Command 1112 of 2416 (84 per node) ran on node nid00022 core 12. Command 12 of 24 (8 per node) ran on node nid00022 core 15.c304-005.ls6.tacc.utexas.edu core 11 Command 13 of 2416 (84 per node) ran on node nid00022c304-006.ls6.tacc.utexas.edu core 13.12 Command 14 of 2416 (84 per node) ran on node nid00022c304-006.ls6.tacc.utexas.edu core 11.13 Command 15 of 2416 (84 per node) ran on node nid00022c304-006.ls6.tacc.utexas.edu core 8.14 Command 16 of 2416 (84 per node) ran on node nid00022c304-006.ls6.tacc.utexas.edu core 9.15 Command 172 of 2416 (84 per node) ran on node nid00021c303-005.ls6.tacc.utexas.edu core 4.1 Command 183 of 2416 (84 per node) ran on node nid00021c303-005.ls6.tacc.utexas.edu core 2. Command 194 of 2416 (84 per node) ran on node nid00021c303-005.ls6.tacc.utexas.edu core 5.3 Command 205 of 2416 (84 per node) ran on node nid00021c303-006.ls6.tacc.utexas.edu core 0.4 Command 216 of 2416 (84 per node) ran on node nid00021c303-006.ls6.tacc.utexas.edu core 3.5 Command 227 of 2416 (84 per node) ran on node nid00021c303-006.ls6.tacc.utexas.edu core 1.6 Command 238 of 2416 (84 per node) ran on node nid00021c303-006.ls6.tacc.utexas.edu core 6.7 Command 249 of 2416 (84 per node) ran on node nid00021c304-005.ls6.tacc.utexas.edu core 7.8 |
Notice that there are 3 4 different host names, each of which ran 8 tasks. This expression:
| Code Block | ||
|---|---|---|
| ||
cat cmd*log | awk '{print $11}' | sort | uniq -c |
should produce output something like this output (read more about piping commands to make a histogram)
| Code Block | ||
|---|---|---|
| ||
4 c302-005.ls6.tacc.utexas.edu 8 nid000214 c302-006.ls6.tacc.utexas.edu 8 nid000224 c305-005.ls6.tacc.utexas.edu 8 nid000234 c305-006.ls6.tacc.utexas.edu |
Some best practices
Redirect task output and error streams
We've already touched on the need to redirect standard output and standard error for each task. Just remember that funny redirection syntax:
| Code Block | ||||
|---|---|---|---|---|
| ||||
my_program input_file1 output_file1 > file1.log 2>&1 |
...
Another really good way to work is to "bundle" a complex set of steps into a shell script that sets up its own environment, loads its own modules, then executes a series of program steps. You can then just call that script, probably with data-specific arguments, in your commands file. This multi-program script is sometimes termed a pipeline, although complex pipelines may involve several such scripts.
For example, you might have a script called align_bwa.sh (a bash script) or align_bowtie2.py (written in python Python) that performs multiple steps needed during the alignment process:
...
The BioITeam maintains a set of such scripts in the /work/projects/BioITeam/common/script directory. Take a look at some of them after you feel more comfortable with initial NGS processing steps. They can be executed by anyone with a TACC account.
...
You may have noticed that all the files involved in our job were in one directory – the batch submissions file, commands file, log files our tasks wrote, and the launcher job output and error files. Of course you'll probably need input files too ![]() as well as output files
as well as output files ![]() .
.
Because a single job can create a lot of files, it is a good idea to use a different directory for each job or set of closely related jobs, maybe with a name similar to the job being performed. This will help you stay organized.
...
As we have seen, there are several special "directory names" the bash shell understands:
- "dot directory" ( . ) refers to "here" or "the current directory"
- "dot dot directory" ( .. ) refers to "one directory up"
- "tilde directory" ( ~ ) refers to your home Home directory
Try these relative path examples:
| Code Block | ||||
|---|---|---|---|---|
| ||||
# navigate through the symbolic link in your Home directory cd $SCRATCH~scratch/core_ngs/slurm/simple ls ../wayness ls ../.. ls -l ~/.bashrc |
(Read more about Absolute and relative pathname syntax)
Interactive sessions (idev)
...
| Code Block | ||||
|---|---|---|---|---|
| ||||
idev -pm development60 -mN 201 -A UT-2015-05-18OTH21164 -Np 1normal -nr 24CoreNGS-Tue |
Notes:
- -p normal requests nodes on the normal queue
- this is the default for our reservation, while
- the development queue is the normal default
- -m 20 60 asks for a 20-60 minute session (120 minutes is the maximum for development)
- -A UT-2015-05-18 OTH21164 specifies the TACC allocation/project to use
- -N 1 asks for 1 node and -n 24 requests access to 24 cores
- --reservation=CoreNGS-Tue gives us priority access to TACC nodes for the class. You normally won't use this option.
When you ask for an idev session, you'll see output as shown below. Note that the process may pause while it waits for available nodes.repeat the "job status: PD" (pending) step while it waits for an available node.
| Code Block |
|---|
-> Checking on the status of development queue. OK
|
| Code Block |
-> Defaults file : ~/.idevrc -> System : ls5ls6 -> Queue : development (cmd line: -p ) -> Nodes : 1 (cmd line: -N ) -> TotalTasks tasksper Node : 24128 (cmd line: -nQueue default ) -> Time (minutes) : 2060 (cmd line: -m ) -> Project : UT-2015-05-1OTH21164 (cmd line: -A ) ----------------------------------------------------------------- Welcome to the Lonestar 5Lonestar6 Supercomputer ----------------------------------------------------------------- No reservation for this job --> Verifying valid submit host (login2)...OK --> Verifying valid jobname...OK --> Enforcing max jobs per user...OK --> Verifying availability of your home dir (/home1/01063/abattenh)...OK-- --> Verifying availabilityvalid ofsubmit your workhost dir (/work/01063/abattenh/lonestar(login1)...OK --> Verifying availability of your scratch dir (/scratch/01063/abattenh)valid jobname...OK --> Verifying valid ssh keys...OK --> Verifying access to desired queue (development)...OK --> Verifying job request isaccess withinto currentdesired queue limits(development)...OK --> Checking available allocation (UT-2015-05-18OTH21164)...OK Submitted batch job 1579644235465 -> After your idev job begins to run, a command prompt will appear, -> and you can begin your interactive development session. -> We will report the job status every 4 seconds: (PD=pending, R=running). ->job> job status: PD ->job> job status: R -> Job is now running on masternode= nid00011c302-005...OK -> Sleeping for 7 seconds...OK -> Checking to make sure your job has initialized an env for you....OK -> Creating interactive terminal session (login) on master node nid00011c302-005. Warning: Permanently added '[nid00011]:6999,[10.128.0.12]:6999' (RSA) to the list of known hosts -> ssh -Y -o "StrictHostKeyChecking no" c302-005 |
Once the idev session has started, it looks quite similar to a login node environment, except for these differences:
- the hostname command on a login node will return a login server name like login2 login3.ls6.tacc.utexas.edu
- while in an idev session hostname returns a compute node name like nid00011 c303-006.ls6.tacc.utexas.edu
- you cannot submit a batch job from inside an idev session, only from a login node
- your idev session will end when the requested time has expired
- or you can just type exit to return to a login node session
...