Objectives
In this lab, you will explore a popular new transcriptome-aware mapper called HISAT2. Simulated RNA-seq data will be provided to you; the data contains paired-end reads that have been generated in silico to replicate real gene count data from Drosophila. The data simulates two biological groups with three biological replicates per group (6 samples total). The objectives of this lab is to:
- Learn how HISAT2 works and how to use it.
- Learn how it is different from using a mapper like BWA.
12 raw data files have been provided for all our further RNA-seq analysis:
- c1_r1, c1_r2, c1_r3 from the first biological condition
- c2_r1, c2_r2, and c2_r3 from the second biological condition
Introduction
HISAT2 is the fastest spliced mapper currently available. It is part of the new tuxedo suite of tools and it will map RNA-Seq data to the genome as well as identify splice junctions. HISAT2, like BWA and bowtie, uses burrows-wheeler transform (BWT) to compress genomes such that they require very little memory to store. Like BWA and bowtie, it builds indexes out of the transformed genomes using a special scheme called FM indexing. This makes it possible to search through these genomes rapidly. Unlike BWA and bowtie, HISAT2 builds a whole genome global index and tens of thousands of small local indexes to make spliced alignment possible. Despite the many indexes, because it uses BWT and FM indexing, the indexes take a very small memory footprint (~5gb RAM for the whole human genome), making it possible to run hisat2 on a standard laptop.
With the human genome, for example, hisat2 builds one global index and 48000 local indexes (each 64000bp long). The size of the local indexes is large enough that 90% of introns will fall into a single local index (on average, human introns are >6kb long).
First, the longer part of a read that maps to the genome contiguously (called the anchor) is mapped using the global index. Once this is mapped, this helps to to identify the relevant local index. HISAT can usually align the remaining part of the read (small anchor) within a single local index rather than searching across the whole genome.
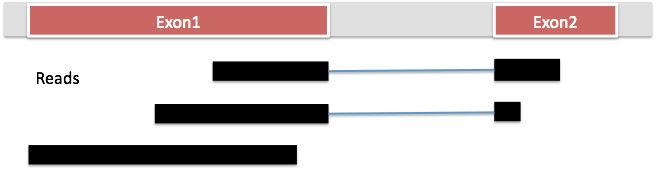
Run HISAT2
First, make sure you are in the right directory for this exercise.
cds cd my_rnaseq_course cd day_1_partB/hisat_exercise ls
Next, see if HISAT2 is a module that is available on stampede.
module spider hisat
As you can see, HISAT2 is not currently a module on stampede. When a program is not available on stampede, you can install it locally in your home or work directory. I have already installed HISAT in my work directory and since my work directory is in your path, you should be able to run hisat2. Type in hisat2 to see if it is in your path and to get usage information.
hisat2
Part 1. Create a index of your reference
NO NEED TO RUN THIS NOW- YOUR INDEX HAS ALREADY BEEN BUILT!
hisat2-build reference/genome.fa reference/genome.fa
Part 2. Align the samples to reference using hisat2
Submit to the TACC queue or run in an idev shell
Create a commands file and use launcher_creator.py followed by sbatch.
nano commands.hisat2 hisat2 -x ../reference/genome.fa -1 ../data/GSM794483_C1_R1_1.fq -2 ../data/GSM794483_C1_R1_2.fq -S GSM794483_C1.sam --phred33 --novel-splicesite-outfile GSM794483_C1.junctions --rna-strandness RF --dta -t hisat2 -x ../reference/genome.fa -1 ../data/GSM794484_C1_R2_1.fq -2 ../data/GSM794484_C1_R2_2.fq -S GSM794484_C1.sam --phred33 --novel-splicesite-outfile GSM794484_C1.junctions --rna-strandness RF --dta -t hisat2 -x ../reference/genome.fa -1 ../data/GSM794485_C1_R3_1.fq -2 ../data/GSM794485_C1_R3_2.fq -S GSM794485_C1.sam --phred33 --novel-splicesite-outfile GSM794485_C1.junctions --rna-strandness RF --dta -t hisat2 -x ../reference/genome.fa -1 ../data/GSM794486_C2_R1_1.fq -2 ../data/GSM794486_C2_R1_2.fq -S GSM794486_C1.sam --phred33 --novel-splicesite-outfile GSM794486_C1.junctions --rna-strandness RF --dta -t hisat2 -x ../reference/genome.fa -1 ../data/GSM794487_C2_R2_1.fq -2 ../data/GSM794487_C2_R2_2.fq -S GSM794487_C1.sam --phred33 --novel-splicesite outfile GSM794487_C1.junctions --rna-strandness RF --dta -t hisat2 -x ../reference/genome.fa -1 ../data/GSM794488_C2_R3_1.fq -2 ../data/GSM794488_C2_R3_2.fq -S GSM794488_C1.sam --phred33 --novel-splicesite-outfile GSM794488_C1.junctions --rna-strandness RF --dta -t
Hisat2 output
1.SAM file : HISAT2 alignment output in standard SAM format.
2.Junctions file : File containing all detected junctions with the format:
chr startpos endpos orientation
3. Log file : The log file should contain alignment summaries.
ls results head results/GSM794483_C1.sam head results/GSM794483_C1.junctions
11607353 reads; of these:
11607353 (100.00%) were paired; of these:
21592 (0.19%) aligned concordantly 0 times
11417720 (98.37%) aligned concordantly exactly 1 time
168041 (1.45%) aligned concordantly >1 times
----
21592 pairs aligned concordantly 0 times; of these:
82 (0.38%) aligned discordantly 1 time
----
21510 pairs aligned 0 times concordantly or discordantly; of these:
43020 mates make up the pairs; of these:
25009 (58.13%) aligned 0 times
9694 (22.53%) aligned exactly 1 time
8317 (19.33%) aligned >1 times
99.89% overall alignment rate