File systems at TACC
The first thing you'll want to do is transfer your sequencing data to TACC so you can process it there. Here is an overview of the different storage areas at TACC, their characteristics, and Linux commands generally used to perform the data transfers:
- wget – retrieves the contents of an Internet URL
- cp – copies files located on any local file system
- scp – copies files to/from a remote system
- rsync – copies directories or files on either local or remote systems
| TACC storage areas and Linux commands to access data (all commands to be executed at TACC except laptop-to-TACC copies, which must be executed on your laptop) |
|
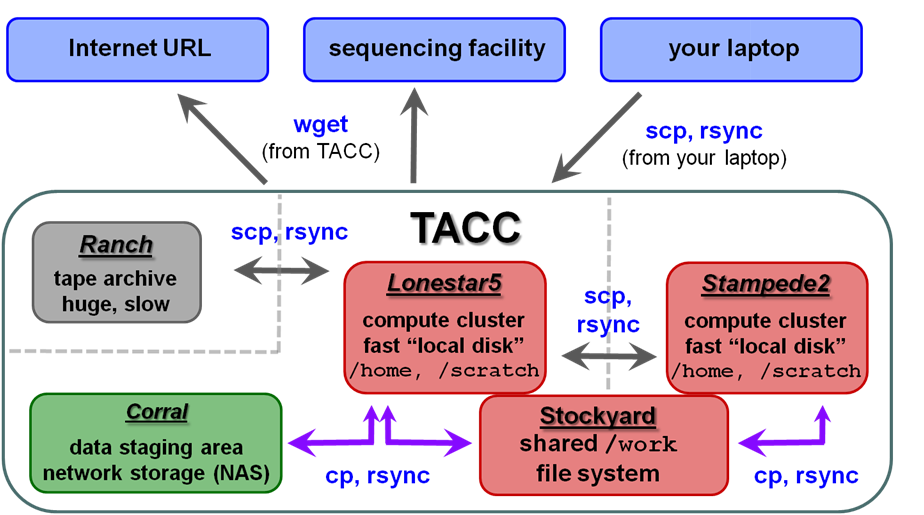
Local file systems
There are 3 local file systems available on any TACC compute cluster (Lonestar5, stampede2, etc.), each with different characteristics. All these local file systems are very fast and set up for parallel I/O (Lustre file system).
On ls5 these local file systems have the following characteristics:
| Home | Work | Scratch | |
|---|---|---|---|
| quota | 10 GB | 1024 GB = 1 TB | 2+ PB (basically infinite) |
| policy | backed up | not backed up, not purged | not backed up, purged if not accessed recently (~10 days) |
| access command | cd | cdw | cds |
| environment variable | $HOME | $STOCKYARD (root of the shared Work file system) $WORK (different sub-directory for each cluster) | $SCRATCH |
| root file system | /home | /work | /scratch |
| use for | Small files such as scripts that you don't want to lose. | Medium-sized artifacts you don't want to copy over all the time. For example, custom programs you install (these can get large), or annotation file used for analysis. | Large files accessed from batch jobs. Your starting files will be copied here from somewhere else, and your final results files will be copied elsewhere (e.g. stockyard, corral, or your BRCF POD). |
When you first login, the system gives you information about disk quota and your compute allocation quota:
--------------------- Project balances for user abattenh ---------------------- | Name Avail SUs Expires | Name Avail SUs Expires | | genomeAnalysis 673 2021-03-31 | BioinformaticsResour 100 2020-06-30 | | UT-2015-05-18 1000 2021-03-31 | DNAdenovo 4969 2021-03-31 | | CancerGenetics 4856 2020-09-30 | A-cm10 8867 2020-12-31 | ------------------------ Disk quotas for user abattenh ------------------------ | Disk Usage (GB) Limit %Used File Usage Limit %Used | | /home1 0.0 10.0 0.10 153 1000000 0.02 | | /work 614.5 1024.0 60.01 61094 3000000 2.04 | | /scratch 2676.6 0.0 0.00 32442 0 0.00 | -------------------------------------------------------------------------------
changing TACC file systems
When you first login, you start in your home directory. Use these commands to change to your other file systems. Notice how your command prompt helpfully changes to show your location.
cdw cds cd
The cd (change directory) command with no arguments takes you to your home directory on any Linux/Unix system. The cdw and cds commands are specific to the TACC environment.
Stockyard (shared Work)
TACC compute clusters now share a common Work file system called stockyard. So files in your Work area do not have to be copied, for example from ls5 to stampede2 – they can be accessed directly from either cluster.
Note that there are two environment variables pertaining to the shared Work area:
- $STOCKYARD - This refers to the root of your shared Work area
- e.g. /work/01063/abattenh
- $WORK - Refers to a sub-directory of the shared Work area that is different for different clusters, e.g.:
- /work/01063/abattenh/lonestar on ls5
- /work/01063/abattenh/stampede2 on stampede2
A mechanism for purchasing larger stockyard allocations (above the 1 TB basic quota) from TACC are in development.
The UT Austin BioInformatics Team, a loose group of bioinformatics researchers, maintains a common directory area on stockyard.
ls /work/projects/BioITeam
Files we will use in this course are in a sub-directory there. The $CORENGS environment variable set in your login profile refers to this path.
echo $CORENGS ls /work/projects/BioITeam/projects/courses/Core_NGS_Tools
Corral
corral is a gigantic (multiple PB) storage system (spinning disk) where researchers can store data. UT researchers may request up to 5 TB of corral storage through the normal TACC allocation request process. Additional space on corral can be rented for ~$85/TB/year.
The UT/Austin BioInformatics Team also has an older, common directory area on corral.
ls /corral-repl/utexas/BioITeam
A couple of things to keep in mind regarding corral:
- corral is a great place to store data in between analyses.
- Store your permanent, original sequence data on corral
- Copy the data you want to work with from corral to $SCRATCH
- Run your analyses (batch jobs)
- Copy your results back to corral
- Occasionally corral can become unavailable. This can cause any command to hang that tries to access corral data!
Ranch
ranch is a gigantic (multiple PB) tape archive system where researchers can archive data. All TACC users have an automatice 2 TB ranch allocation. UT researchers may request larger (multi-TB) ranch storage allocations through the normal TACC allocation request process.
There is currently no charge for ranch storage. However, since the data is stored on tape it is not immediately available – robots find and mount appropriate tapes when the data is requested, and it can take minutes to hours for the data to appear on disk. The metadata about your data – the directory structures and file names – is always accessible, but the actual data in the files is not on disk until "staged". See the ranch user guide for more information: https://www.tacc.utexas.edu/user-services/user-guides/ranch-user-guide.
Once that data is staged to the ranch disk it can be copied to other places. However, the ranch file system is not mounted as a local file system from the stampede2 or ls5 clusters. So remote copy commands are always needed to copy data to and from ranch (e.g. scp, rsync).
Staging your data
So, your sequencing center has some data for you. They may send you a list of web or FTP links to use to download the data.
The first task is to get this sequencing data to a permanent storage area. This should not be your laptop! corral (or stockyard) is a great place for it, or a server maintained by your lab or company.
Here's an example of a "best practice". Wherever your permanent storage area is, it should have a rational sub-directory structure that reflects its contents. It's easy to process a few NGS datasets, but when they start multiplying like tribbles, good organization and naming conventions will be the only thing standing between you and utter chaos!
For example:
- original – for original sequencing data (compressed FASTQ files)
- sub-directories named, for example, by year_month.<project_name>
- aligned – for alignment artifacts (BAM files, etc)
- sub-directories named, e.g., by year_month.<project_name>
- analysis – further downstream analysis
- reasonably named sub-directories, often by project
- genome – reference genomes and other annotation files used in alignment and analysis
- sub-directories for different reference genomes
- e.g. ucsc/hg19, ucsc/sacCer3, mirbase/v20
- code – for scripts and programs you and others in your organization write
- ideally maintained in a version control system such as git, subversion or cvs.
- easiest to name sub-directories for people.
Download from a link – wget
Well, you don't have a desktop at TACC to "Save as" to, so what to do with a link? The wget program knows how to access web URLs such as http, https and ftp.
wget
Get ready to run wget from the directory where you want to put the data.
Don't press Enter after the wget command – just put a space after it.
mkdir -p $WORK/archive/original/2020.core_ngs cd $WORK/archive/original/2020.core_ngs wget
Here are two web links:
- http://web.corral.tacc.utexas.edu/BioITeam/yeast_stuff/Sample_Yeast_L005_R1.cat.fastq.gz
- http://web.corral.tacc.utexas.edu/BioITeam/yeast_stuff/Sample_Yeast_L005_R2.cat.fastq.gz
Right-click (Windows) or Control+click (Mac) on the 1st link in your browser, then select "Copy link location" from the menu. Now go back to your Terminal. Put your cursor after the space following the wget command then either right-click (Windows), or Paste (Command-V on Mac, Control-V on Windows). The command line to be executed should now look like this:
wget http://web.corral.tacc.utexas.edu/BioITeam/yeast_stuff/Sample_Yeast_L005_R1.cat.fastq.gz
Now press Enter to get the command going. Repeat for the 2nd link. Check that you now see the two files (ls).
Copy from a corral location - cp or rsync
Suppose you have a corral allocation or stockyard area where your organization keeps its data, and that the sequencing data has been downloaded there. You can use various Linux commands to copy the data locally from there to your $SCRATCH area.
cp
The cp command copies one or more files from a local source to a local destination. It has the most common form:
cp [options] <source file 1> <source file 2> ... <destination directory>/
Make a directory in your scratch area and copy a single file to it. The trailing slash ( / ) on the destination says it is a directory.
mkdir -p $SCRATCH/data/test1 cp $CORENGS/misc/small.fq $SCRATCH/data/test1/ ls $SCRATCH/data/test1 # or.. mkdir -p ~/scratch/data/test1 cd ~/scratch/data/test1 cp $CORENGS/misc/small.fq . ls
Copy an entire directory to your scratch area. The -r argument says "recursive".
mkdir -p $SCRATCH/data cds cd data cp -r $CORENGS/general/ general/
Exercise: What files were copied over?
local rsync
The rsync command is typically used to copy whole directories. What's great about rsync is that it only copies what has changed in the source directory. So if you regularly rsync a large directory to TACC, it may take a long time the 1st time, but the 2nd time (say after downloading more sequencing data to the source), only the new files will be copied.
rsync is a very complicated program, with many options (http://rsync.samba.org/ftp/rsync/rsync.html). However, if you use the recipe shown here for directories, it's hard to go wrong:
rsync -avW local/path/to/source_directory/ local/path/to/destination_directory/
Both the source and target directories are local (in some file system accessible directly from ls5). Either full or relative path syntax can be used for both. The -avP options above stand for:
- -a means "archive mode", which implies the following options (and a few others)
- -p – preserve file permissions
- -t – preserve file times
- -l – copy symbolic links as links
- -r – recursively copy sub-directories
- -v means verbose
- -W means transfer Whole file only
- Normally the rsync algorithm compares the contents of files that need to be copied and only transfers the different parts.
- For large files and binary files, figuring out what has changed (diff-ing) can take more time than just copying the whole file.
- The -W option disables file content comparisons (skips diff-ing).
Since these are all single-character options, they can be combined after one option prefix dash ( - ). You could also use options -ptlrvW, separately, instead of using -a for "archive mode".
Always add a trailing slash ( / ) after directory names
The trailing slash ( / ) on the source and destination directories are very important for rsync (and for other Linux copy commands also)!
rsync will create the last directory level for you, but earlier levels must already exist.
mkdir -p $SCRATCH/data cds rsync -avWP $CORENGS/custom_tracks/ data/custom_tracks/
Exercise: What files were copied over?
Now repeat the rsync and see the difference.
Use the Up arrow to retrieve the previous command from your bash command history.
rsync -avWP /work/projects/BioITeam/projects/courses/Core_NGS_Tools/custom_tracks/ data/custom_tracks/
The bash shell has several convenient line editing features:
- use the Up arrow to scroll back through the command line history; Down arrow goes forward
- use Ctrl-a to move the cursor to the beginning of a line; Ctrl-e to the end
- use Backspace to remove text before the cursor; Delete to remove text after the cursor
Copy from a remote computer - scp or rsync
Provided that the remote computer is running Linux and you have ssh access to it, you can use various Linux commands to copy data over a secure connection.
The good news is that once you have learned cp and local rsync, remote secure copy (scp) and remote rsync are very similar!
scp
The scp command copies one or more files from a source to a destination, where either source or destination, or both, can be a remote path.
Remote paths are similar to local paths, but have user and host information first:
user_name@full.host.name:/full/path/to/directory/or/file
– or –
user_name@full.host.name:~/path/relative/to/home/directory
Copy a single file to your $SCRATCH/data/test1 directory from the server named gapdh.icmb.utexas.edu, using the user account corengstools. When prompted for a password, use the one we have written on the board (or copy/paste the password from this file: $CORENGS/tacc/gapdh_access.txt)
cds cat $CORENGS/tacc/gapdh_access.txt scp corengstools@gapdh.icmb.utexas.edu:~/custom_tracks/progeria_ctcf.vcf.gz ./data/test1/ ls ./data/test1
Notes:
- The 1st time you access a new host the SSH security prompt will appear
- You will be prompted for your remote host password
- The -r recursive argument works for scp also, just like for cp
remote rsync
rsync can be run just like before, but using the remote-host syntax. Here we use two tricks:
- The tilde ( ~ ) at the start of the path means "relative to my home directory"
- We traverse through the BioITeam symbolic link created in your home directory earlier.
- We use the same tilde ( ~ ) in the destination to traverse the scratch symbolic link in your home directory.
Don't forget to change userid below.
rsync -ptlrvP corengstools@gapdh.icmb.utexas.edu:~/custom_tracks/ ~/scratch/data/custom_tracks/
Exercise: Was anything copied?
Scavenger hunt exercise
Here's a fun scavenger hunt for more practice. Issue the following commands to get practice what you've learned so far:
To get started:
cd cp -r $CORENGS/linuxpractice/what what cd what cat readme
Where are you when you're all done?