Setup
Logon and idev
First login to ls6 like you did before. Then start an idev session so that we don't do too much processing on the login nodes.
idev -m 120 -N 1 -A OTH21164 -r CoreNGS-Tue # -or- idev -m 90 -N 1 -A OTH21164 -p development
Data staging
Set ourselves up to process some yeast data data in $SCRATCH, using some of best practices for organizing our workflow.
# Create a $SCRATCH area to work on data for this course, # with a sub-directory for pre-processing raw fastq files mkdir -p $SCRATCH/core_ngs/fastq_prep # Make symbolic links to the original yeast data: cd $SCRATCH/core_ngs/fastq_prep ln -s -f $CORENGS/yeast_stuff/Sample_Yeast_L005_R1.cat.fastq.gz ln -s -f $CORENGS/yeast_stuff/Sample_Yeast_L005_R2.cat.fastq.gz # or ln -s -f ~/CoreNGS/yeast_stuff/Sample_Yeast_L005_R1.cat.fastq.gz ln -s -f ~/CoreNGS/yeast_stuff/Sample_Yeast_L005_R2.cat.fastq.gz # or ln -sf /work/projects/BioITeam/projects/courses/Core_NGS_Tools/yeast_stuff/Sample_Yeast_L005_R1.cat.fastq.gz ln -sf /work/projects/BioITeam/projects/courses/Core_NGS_Tools/yeast_stuff/Sample_Yeast_L005_R2.cat.fastq.gz
Illumina sequence data format (FASTQ)
GSAF gives you paired end sequencing data in two matching FASTQ format files, containing reads for each end sequenced. See where your data really is and how big it is.
# the -l options says "long listing" which shows where the link goes, # but doesn't show details of the real file ls -l # the -L option says to follow the link to the real file, # -l means long listing (includes size) # -h says "human readable" (e.g. MB, GB) ls -Llh
4-line FASTQ format
Each read end sequenced is represented by a 4-line entry in the FASTQ file that looks like this:
@HWI-ST1097:127:C0W5VACXX:5:1101:4820:2124 1:N:0:CTCAGA TCTCTAGTTTCGATAGATTGCTGATTTGTTTCCTGGTCATTAGTCTCCGTATTTATATTATTATCCTGAGCATCATTGATGGCTGCAGGAGGAGCATTCTC + CCCFFFFDHHHHHGGGHIJIJJIHHJJJHHIGHHIJFHGICA91CGIGB?9EF9DDBFCGIGGIIIID>DCHGCEDH@C@DC?3AB?@B;AB??;=A>3;;
Line 1 is the unique read name. The format for Illumina reads is as follows, using the read name above:
machine_id:lane:flowcell_grid_coordinates end_number:failed_qc:0:barcode
@HWI-ST1097:127:C0W5VACXX:5:1101:4820:2124 1:N:0:CTCAGA
- The line as a whole will be unique for this read fragment.
- the corresponding R1 and R2 reads will have identical machine_id:lane:flowcell_grid_coordinates information. This common part of the name ties the two read ends together
- the end_number:failed_qc:0:barcode information will be different for R1 and R2
- Most sequencing facilities will not give you qc-failed reads (failed_qc = Y) unless you ask for them.
Line 2 is the sequence reported by the machine, starting with the first base of the insert (the 5' adapter has usually been removed by the sequencing facility). These are ACGT or N uppercase characters.
Line 3 always starts with '+' (it can optionally include a sequence description)
Line 4 is a string of Ascii-encoded base quality scores, one character per base in the sequence.
- For each base, an integer Phred-type quality score is calculated as
integer score = -10 log(probabilty base is wrong)then added to 33 to make a number in the Ascii printable character range. - As you can see from the table below, alphabetical letters - good, numbers – ok, most special characters – bad (except :;<=>?@).
- See https://www.asciitable.com
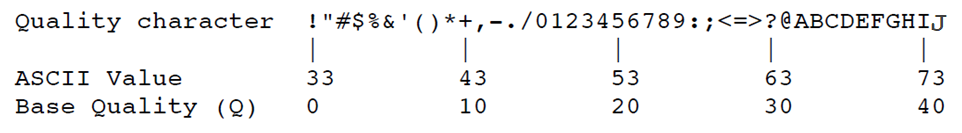
See the Wikipedia FASTQ format page for more information.
Exercise: What character in the quality score string in the FASTQ entry above represents the best base quality? Roughly what is the error probability estimated by the sequencer?
About compressed files
Sequencing data files can be very large - from a few megabytes to gigabytes. And with NGS giving us longer reads and deeper sequencing at decreasing price points, it's not hard to run out of storage space. As a result, most sequencing facilities will give you compressed sequencing data files.
The most common compression program used for individual files is gzip whose compressed files have the .gz extension. The tar and zip programs are most commonly used for compressing directories.
Let's take a look at the size difference between uncompressed and compressed files. We use the -l option of ls to get a long listing that includes the file size, and -h to have that size displayed in "human readable" form rather than in raw byte sizes.
ls -lh $CORENGS/yeast_stuff/*L005*.fastq ls -lh $CORENGS/yeast_stuff/*L005*.fastq.gz
Pathname wildcarding
The asterisk character ( * ) is a pathname wildcard that matches 0 or more characters.
Read more about pathname wildcards: Pathname wildcards
Exercise: About how big are the compressed files? The uncompressed files? About what is the compression factor?
You may be tempted to want to un-compress your sequencing files in order to manipulate them more directly – but resist that temptation! Nearly all modern bioinformatics tools are able to work on .gz files, and there are tools and techniques for working with the contents of compressed files without ever un-compressing them.
gzip and gunzip
With no options, gzip compresses the file you give it in-place. Once all the content has been compressed, the original uncompressed file is removed, leaving only the compressed version (the original file name plus a .gz extension). The gunzip function works in a similar manner, except that its input is a compressed file with a .gz file and produces an uncompressed file without the .gz extension.
# if the $CORENGS environment variable is not defined export CORENGS=/work/projects/BioITeam/projects/courses/Core_NGS_Tools # make sure you're in your $SCRATCH/core_ngs/fastq_prep directory cd $SCRATCH/core_ngs/fastq_prep # Copy over a small, uncompressed fastq file cp $CORENGS/misc/small.fq . # check the size, then compress it in-place ls -lh small* gzip small.fq # check the compressed file size ls -lh small* # uncompress it again gunzip small.fq.gz ls -lh small*
Both gzip and gunzip are extremely I/O intensive when run on large files.
While TACC has tremendous compute resources and its specialized parallel file system is great, it has its limitations. It is not difficult to overwhelm the TACC file system if you gzip or gunzip more than a few files at a time – as few as 5-6!
The intensity of compression/decompression operations is another reason you should compress your sequencing files once (if they aren't already) then leave them that way.
head and tail, more or less
One of the challenges of dealing with large data files, whether compressed or not, is finding your way around the data – finding and looking at relevant pieces of it. Except for the smallest of files, you can't open them up in a text editor because those programs read the whole file into memory, so will choke on sequencing data files! Instead we use various techniques to look at pieces of the files at a time. (Read more about commands for Displaying file contents)
The first technique is the use of pagers – we've already seen this with the more command. Review its use now on our small uncompressed file:
# Use spacebar to advance a page; Ctrl-c to exit more small.fq
Another pager, with additional features, is less. The most useful feature of less is the ability to search – but it still doesn't load the whole file into memory, so searching a really big file can be slow.
Here's a summary of the most common less navigation commands, once the less pager is active. It has tons of other options (try less --help).
- q – quit
- Ctrl-f or space – page forward
- Ctrl-b – page backward
- /<pattern> – search for <pattern> in forward direction
- n – next match
- N – previous match
- ?<pattern> – search for <pattern> in backward direction
- n – previous match going back
- N – next match going forward
If you start less with the -N option, it will display line numbers.
Exercise: What line of small.fq contains the read name with grid coordinates 2316:10009:100563?
head
For a really quick peek at the first few lines of your data, there's nothing like the head command. By default head displays the first 10 lines of data from the file you give it or from its standard input. With an argument -NNN (that is a dash followed by some number), it will show that many lines of data.
# shows 1st 10 lines head small.fq # shows 1st 100 lines -- might want to pipe this to more to see a bit at a time head -100 small.fq | more
So what if you want to see line numbers on your head or tail output? Neither command seems to have an option to do this.
piping
So what is that vertical bar ( | ) all about? It is the pipe operator!
The pipe operator ( | ) connects one program's standard output to the next program's standard input. The power of the Linux command line is due in no small part to the power of piping. (Read more about Piping and Standard Unix I/O streams)
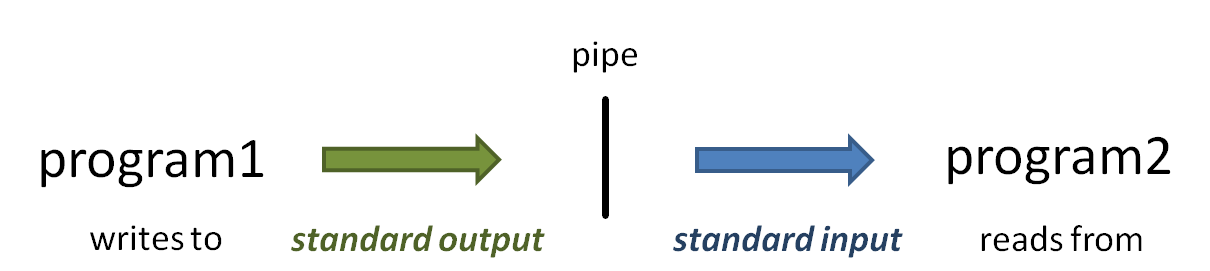
- When you execute the head -100 small.fq | more command, head starts writing lines of the small.fq file to standard output.
- Because of the pipe, the output does not go to the Terminal, but is connected to the standard input of the more command.
- Instead of reading lines from a file you specify as a command-line argument, more obtains its input from standard input.
- The more command writes a page of text to standard output, which is displayed on the Terminal.
Programs often allow input from standard input
Most Linux commands are designed to accept input from standard input in addition to (or instead of) command line arguments so that data can be piped in.
Many bioinformatics programs also allow data to be piped in. Often they will require you provide a special argument, such as stdin or -, to tell the program data is coming from standard input instead of a file.
tail
The yang to head's ying is tail, which by default it displays the last 10 lines of its data, and also uses the -NNN syntax to show the last NNN lines. (Note that with very large files it may take a while for tail to start producing output because it has to read through the file sequentially to get to the end.)
But what's really cool about tail is its -n +NN syntax. This displays all the lines starting at line NN. Note this syntax: the -n option switch follows by a plus sign ( + ) in front of a number – the plus sign is what says "starting at this line"! Try these examples:
# shows the last 10 lines tail small.fq # shows the last 100 lines -- might want to pipe this to more to see a bit at a time tail -100 small.fq | more # shows all the lines starting at line 900 -- better pipe it to a pager! # cat -n adds line numbers to its output so we can see where we are in the file cat -n small.fq | tail -n +900 | more # shows 15 lines starting at line 900 because we pipe to head -15 tail -n +900 small.fq | head -15
zcat and gunzip -c tricks
Ok, now you know how to navigate an un-compressed file using head and tail, more or less. But what if your FASTQ file has been compressed by gzip? You don't want to un-compress the file, remember?
So you use the gunzip -c trick. This un-compresses the file, but instead of writing the un-compressed data to another file (without the .gz extension) it write it to its standard output where it can be piped to programs like your friends head and tail, more or less.
Let's illustrate this using one of the compressed files in your fastq_prep sub-directory:
# make sure you're in your $SCRATCH/core_ngs/fastq_prep directory cd $SCRATCH/core_ngs/fastq_prep gunzip -c Sample_Yeast_L005_R1.cat.fastq.gz | more gunzip -c Sample_Yeast_L005_R1.cat.fastq.gz | head gunzip -c Sample_Yeast_L005_R1.cat.fastq.gz | tail gunzip -c Sample_Yeast_L005_R1.cat.fastq.gz | tail -n +901 | head -8 # Note that less will display .gz file contents automatically less -N Sample_Yeast_L005_R1.cat.fastq.gz
Finally, another command that does the same thing as gunzip -c is zcat – which is like cat except that it works on gzip-compressed (.gz) files!
zcat Sample_Yeast_L005_R1.cat.fastq.gz | more zcat Sample_Yeast_L005_R1.cat.fastq.gz | less -N zcat Sample_Yeast_L005_R1.cat.fastq.gz | head zcat Sample_Yeast_L005_R1.cat.fastq.gz | tail zcat Sample_Yeast_L005_R1.cat.fastq.gz | tail -n +901 | head -8 # include original line numbers zcat Sample_Yeast_L005_R1.cat.fastq.gz | cat -n | tail -n +901 | head -8
There will be times when you forget to pipe your large zcat or gunzip -c output somewhere – even the experienced among us still make this mistake! This leads to pages and pages of data spewing across your Terminal.
If you're lucky you can kill the output with Ctrl-c. But if that doesn't work (and often it doesn't) just close your Terminal window. This terminates the process on the server (like hanging up the phone), then you just can log back in.
Counting your sequences
One of the first thing to check is that your FASTQ files are the same length, and that length is evenly divisible by 4. The wc command (word count) using the -l switch to tell it to count lines, not words, is perfect for this. It's so handy that you'll end up using wc -l a lot to count things. It's especially powerful when used with filename wild carding.
wc -l small.fq head -100 small.fq > small2.fq wc -l small*.fq
You can also pipe the output of zcat or gunzip -c to wc -l to count lines in your compressed FASTQ file.
Exercise: How many lines are in the Sample_Yeast_L005_R1.cat.fastq.gz file? How many sequences is this?
How to do math on the command line
The bash shell has a really strange syntax for arithmetic: it uses a double-parenthesis operator after the $ sign (which means evaluate this expression). Go figure.
echo $((2368720 / 4))
Here's another trick: backticks evaluation. When you enclose a command expression in backtick quotes ( ` ) the enclosed expression is evaluated and its standard output substituted into the string. (Read more about Quoting in the shell)
Here's how you would combine this math expression with zcat line counting on your file using the magic of backtick evaluation. Notice that the wc -l expression is what is reading from standard input.
cd $SCRATCH/core_ngs/fastq_prep zcat Sample_Yeast_L005_R1.cat.fastq.gz | echo "$((`wc -l` / 4))"
Whew!
bash arithmetic is integer valued only
Note that arithmetic in the bash shell is integer valued only, so don't use it for anything that requires decimal places!
A better way to do math
Well, doing math in bash is pretty awful – there has to be something better. There is! It's called awk, which is a powerful scripting language that is easily invoked from the command line.
In the code below we pipe the output from wc -l (number of lines in the FASTQ file) to awk, which executes its body (the statements between the curly braces ( { } ) for each line of input. Here the input is just one line, with one field – the line count. The awk body just divides the 1st input field ($1) by 4 and writes the result to standard output. (Read more about awk in Advanced commands: awk)
cd $SCRATCH/core_ngs/fastq_prep
zcat Sample_Yeast_L005_R1.cat.fastq.gz | wc -l | awk '{print $1 / 4}'
Note that $1 means something different in awk – the 1st whitespace-delimited input field – than it does in bash, where it represents the 1st argument to a script or function (technically, the 1 environment variable). This is an example of where a metacharacter - the dollar sign ( $ ) here – has a different meaning for two different programs.
The bash shell treats dollar sign ( $ ) as an evaluation operator, so will normally attempt to evaluate the environment variable name following the $ and substitute its value in the output (e.g. echo $SCRATCH). But we don't want that evaluation to be applied to the {print $1 / 4} script argument passed to awk; instead we want awk to see the literal string {print $1 / 4} as its script. To achieve this result we surround the script argument with single quotes ( ' ' ), which tells the shell to treat everything enclosed by the quotes as literal text, and not perform any metacharacter evaluation.
(Read more about Quoting in the shell)
Processing multiple compressed files
You've probably figured out by now that you can't easily use filename wildcarding along with zcat and piping to process multiple files. For this, you need to code a for loop in bash. Fortunately, this is pretty easy. Try this:
cd $SCRATCH/core_ngs/fastq_prep
for fname in *.gz; do
echo "Processing $fname"
echo "..$fname has `zcat $fname | wc -l | awk '{print $1 / 4}'` sequences"
done
Each time through the for loop, the next item in the argument list (here the files matching the wildcard glob *.gz) is assigned to the for loop's formal argument (here the variable fname). The actual filename is then referenced as $fname inside the loop. (Read more about Bash control flow)