Introduction
breseq is a tool developed by the Barrick lab intended for analyzing genome re-sequencing data for bacteria. It is primarily used to analyze laboratory evolution experiments with microbes. In these experiments, there is usually a high-quality reference genome for the ancestral strain, and one is interested in exhaustively finding all of the mutations that occurred during the evolution experiment. Then one might want to construct a phylogenetic tree of individuals samples from a single population or determine whether the same gene is mutated in many independent evolution experiments in an environment.
Input data / expectations:
- Haploid reference genome
- Relatively small (<20 Mb) reference genome
- Input FASTQ reads can be from any sequencing technology
- Average genomic coverage > 30-fold
- Less than ~1,000 mutations expected
- Detects SNVs and SVs from single-end reads (does not use paired-end distance information)
- Produces annotated HTML output
You can learn a great deal more about breseq by reading the Online Documentation.
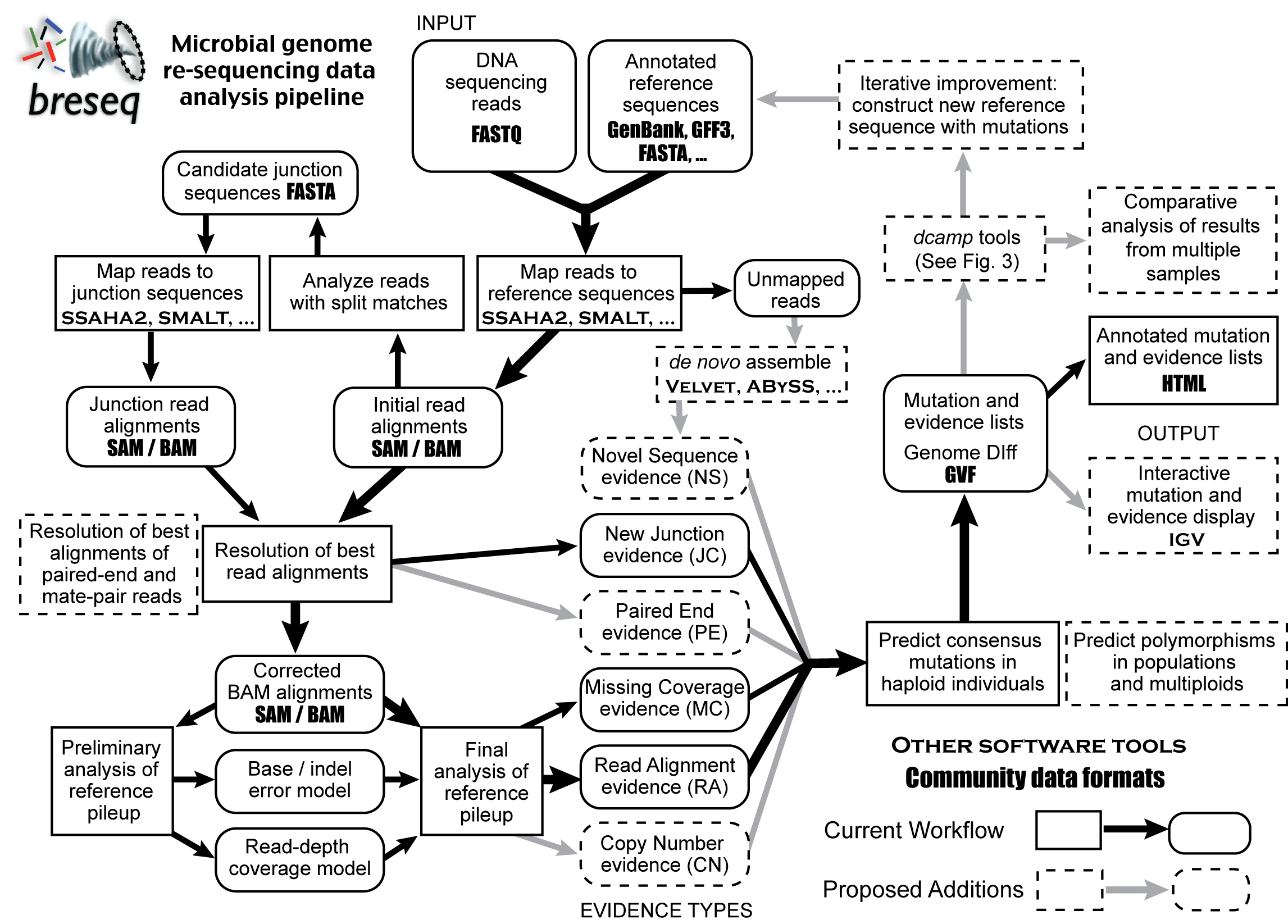
Here is a rough outline of the workflow in breseq with proposed additions.
This tutorial was reformatted from the most recent version found here. Our thanks to the previous instructors.
Objectives:
- Use a very self contained/automated pipeline to identify mutations.
- Explain the types of mutations found in a complete manner before using methods better suited for higher order organisms.
Bacteriophage lambda data set
First, we'll run breseq on a small data set to be sure that it is installed correctly, and to get a taste for what the output looks like. This sample is a mixed population of bacteriophage lambda that was co-evolved in lab with its E. coli hosts.
Environment
In order to run breseq, we need to make sure breseq was made available to you when we set up your .bashrc file on the first day.
tacc:~$ which breseq /corral-repl/utexas/BioITeam/breseq/bin/breseq
breseq should now run using the breseq command. breseq by itself will show you what the command expectations are. Not all programs are configured to tell you what it expects just from typing the name of it. Some require the name of the command followed by one of the following: -h or --help or ? while others require preceding the command name with "man" (short for manual). If all that fails google is your friend for all programs not named "R" ... google is still your best bet, but it won't be your friend. In the specific case of R, adding the word stat somewhere to the search will greatly help things.
Data
The data files for this tutorial is located in following location:
/corral-repl/utexas/BioITeam/ngs_course/lambda_mixed_pop/data/
Copy the contents of this directory to a new directory called BDIB_breseq_lambda_mixed_pop in your scratch directory.
Now use the ls command to see what files were copied:
File Name | Description | Sample |
|---|---|---|
| Single-end Illumina 36-bp reads | Evolved lambda bacteriophage mixed population genome sequencing |
| Reference Genome | Bacteriophage lambda |
Running breseq
Because this data set is relatively small (roughly 100x coverage of a 48,000 bp genome), a breseq run will take < 5 minutes, but it is computationally intense enough that it should not be run on the head node. By now this should be somewhat familiar, but incase its not expand the following.
cd $SCRATCH/BDIB_breseq_lambda_mixed_pop module unload samtools breseq -j 48 -r lambda.gbk lambda_mixed_population.fastq &> log.txt &
A bunch of progress messages will stream by during the breseq run which would clutter the screen if not for the redirection to the log.txt file. The & at the end of the line tells the system to run the previous command in the background which will enable you to still type and execute other commands while breseq runs. The output text details several steps in a pipeline that combines the steps of mapping (using SSAHA2), variant calling, annotating mutations, etc. You can examine them by peeking in the log.txt file as your job runs using tail log.txt. While breseq is running lets look at what the different parts of the command are actually doing:
| part | puprose |
|---|---|
| -j 48 | Use 48 processors (the max available on lonestar5 nodes) |
| -r lambda.gbk | Use the lambda.gbk file as the reference to identify specific mutations |
| lambda_mixed_population.fastq | breseq assumes any argument not preceded by a - option to be an input fastq file to be used for mapping |
| &> log.txt | redirect the output and error to the file log.txt |
| & | run the preceding command in the background |
This will finish very quickly (likely before you begin reading this) with a final line of "Creating index HTML table...". check this using the tail command. If you instead see this output:
!!!!!!!!!!!!!!!!!!!!!!!> FATAL ERROR <!!!!!!!!!!!!!!!!!!!!!!! Error running command: [system] /opt/apps/samtools/1.3/bin/samtools sort ./03_candidate_junctions/best.unsorted.bam ./03_candidate_junctions/best Result code: 256 FILE: libbreseq/common.h LINE: 1294 !!!!!!!!!!!!!!!!!!!!!!!> FATAL ERROR <!!!!!!!!!!!!!!!!!!!!!!! breseq: libbreseq/common.h:92: void breseq::my_assertion_handler(bool, const char*, const char*, int, const string&): Assertion `false' failed. Aborted
It means that you tried to run breseq with the incorrect version of samtools loaded. Execute the following 4 commands and then retry running breseq.
Looking at breseq predictions
breseq produced a lot of directories beginning 01_sequence_conversion, 02_reference_alignment, ... Each of these contains intermediate files that are used to 'pickup where it left off' if the run doesn't complete successfully. These can be deleted when the run completes, or explored if you are interested in the inner guts of what is going on. More importantly, breseq will also produce two directories called: data and output which contain files used to create .html output files and .html output files respectively. The most interesting files are the .html files which can't be viewed directly on lonestar. Therefore we first need to copy the output directory back to your desktop computer. Use scp to transfer the contents of the output directory back to your local computer.
Navigate to the output directory in the finder and open the a file called index.html. This will open the results in a web browser window that you can click through different mutations and other information and see the evidence supporting it. The summary page provides useful information about the percent of reads mapping to the genome as well as the overall coverage of the genome. The Mutation Predictions page is where most of the analysis time is spent in determining which mutations are important (and more rarely inaccurate).
Click around through the different mutations and examine their evidence to see what kinds of mutations you can identify. Interact with your instructors, and show us what different types of mutations you are able to identify, or ask us what mutations you don't understand. Additional information on analyzing the output can be found at the following reference:
- Deatherage, D.E., Barrick, J.E.. (2014) Identification of mutations in laboratory-evolved microbes from next-generation sequencing data using breseq. Methods Mol. Biol. 1151:165-188. «PubMed»